Tidy Transcriptomics For Single-Cell RNA Sequencing Analyses
Stefano Mangiola, Walter and Eliza Hall Institute1
Maria Doyle, Peter MacCallum Cancer Centre2
Source:vignettes/tidytranscriptomics_case_study.Rmd
tidytranscriptomics_case_study.RmdWorkshop introduction


Instructors
Dr. Stefano Mangiola is currently a Postdoctoral researcher in the laboratory of Prof. Tony Papenfuss at the Walter and Eliza Hall Institute in Melbourne, Australia. His background spans from biotechnology to bioinformatics and biostatistics. His research focuses on prostate and breast tumour microenvironment, the development of statistical models for the analysis of RNA sequencing data, and data analysis and visualisation interfaces.
Dr. Maria Doyle is the Application and Training Specialist for Research Computing at the Peter MacCallum Cancer Centre in Melbourne, Australia. She has a PhD in Molecular Biology and currently works in bioinformatics and data science education and training. She is passionate about supporting researchers, reproducible research, open source and tidy data.
Description
This tutorial will showcase analysis of single-cell RNA sequencing data following the tidy data paradigm. The tidy data paradigm provides a standard way to organise data values within a dataset, where each variable is a column, each observation is a row, and data is manipulated using an easy-to-understand vocabulary. Most importantly, the data structure remains consistent across manipulation and analysis functions.
This can be achieved with the integration of packages present in the R CRAN and Bioconductor ecosystem, including tidySingleCellExperiment, tidySummarizedExperiment, tidybulk and tidyverse. These packages are part of the tidytranscriptomics suite that introduces a tidy approach to RNA sequencing data representation and analysis. For more information see the tidy transcriptomics blog.
Goals and objectives
- To approach single-cell data representation and analysis through a tidy data paradigm, integrating tidyverse with tidySingleCellExperiment.
- Compare SingleCellExperiment and tidy representation
- Apply tidy functions to SingleCellExperiment objects
- Reproduce a real-world case study that showcases the power of tidy single-cell methods
What you will learn
- Basic
tidyoperations possible withtidySingleCellExperiment - The differences between
SingleCellExperimentrepresentation andtidyrepresentation - How to interface
SingleCellExperimentwith tidy manipulation and visualisation - A real-world case study that will showcase the power of
tidysingle-cell methods compared with base/ad-hoc methods
What you will not learn
- The molecular technology of single-cell sequencing
- The fundamentals of single-cell data analysis
- The fundamentals of tidy data analysis
This workshop will demonstrate a real-world example of using tidy transcriptomics packages to analyse single cell data. This workshop is not a step-by-step introduction in how to perform single-cell analysis. For an overview of single-cell analysis steps performed in a tidy way please see the ISMB2021 workshop.
Getting started
Cloud
Easiest way to run this material. We will use the Orchestra Cloud platform during the BioC2022 workshop.
- Go to Orchestra.
- Log in.
- Find the workshop. In the search box type bioc2022, sort by Created column, and select the most recently created workshop called “BioC2022: Tidy Transcriptomics For Single-Cell RNA Sequencing Analyses” There are several tidy transcriptomics workshops. Be sure to select the BioC2022 one with the most recent created date.
- Click “Launch” (may take a minute or two).
- Follow instructions.. Do not share your personalized URL for the RStudio session, or use the trainers, as only one browser at a time can be connected.
- Open
tidytranscriptomics_case_study.Rmdinbioc2022_tidytranscriptomcs/vignettesfolder
Slides
The embedded slides below may take a minute to appear. You can also view or download here.
Part 1 Introduction to tidySingleCellExperiment
# Load packages
library(SingleCellExperiment)
library(ggplot2)
library(plotly)
library(dplyr)
library(colorspace)
library(dittoSeq)SingleCellExperiment is a very popular analysis toolkit for single cell RNA sequencing data (Butler et al. 2018; Stuart et al. 2019).
Here we load single-cell data in SingleCellExperiment object format. This data is peripheral blood mononuclear cells (PBMCs) from metastatic breast cancer patients.
# load single cell RNA sequencing data
sce_obj <- bioc2022tidytranscriptomics::sce_obj
# take a look
sce_obj## class: SingleCellExperiment
## dim: 482 3580
## metadata(0):
## assays(2): counts logcounts
## rownames(482): HBB HBA2 ... TRGC2 CD8A
## rowData names(0):
## colnames(3580): 1_AAATGGACAAGTTCGT-1 1_AACAAGAGTGTTGAGG-1 ...
## 10_TTTGTTGGTACACGTT-1 10_TTTGTTGGTGATAGAT-1
## colData names(15): Barcode batch ... treatment sample
## reducedDimNames(1): UMAP
## mainExpName: SCT
## altExpNames(1): RNAtidySingleCellExperiment provides a bridge between the SingleCellExperiment single-cell package and the tidyverse (Wickham et al. 2019). It creates an invisible layer that enables viewing the SingleCellExperiment object as a tidyverse tibble, and provides SingleCellExperiment-compatible dplyr, tidyr, ggplot and plotly functions.
If we load the tidySingleCellExperiment package and then view the single cell data, it now displays as a tibble.
library(tidySingleCellExperiment)
sce_obj## # A SingleCellExperiment-tibble abstraction: 3,580 × 18
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell Barcode batch BCB S.Score G2M.S…¹ Phase cell_…² nCoun…³ nFeat…⁴
## <chr> <fct> <fct> <fct> <dbl> <dbl> <fct> <fct> <int> <int>
## 1 1_AAATGGA… AAATGG… 1 BCB1… -2.07e-2 -0.0602 G1 TCR_V_… 215 36
## 2 1_AACAAGA… AACAAG… 1 BCB1… 2.09e-2 -0.0357 S CD8+_t… 145 41
## 3 1_AACGTCA… AACGTC… 1 BCB1… -2.54e-2 -0.133 G1 CD8+_h… 356 44
## 4 1_AACTTCT… AACTTC… 1 BCB1… 2.85e-2 -0.163 S CD8+_t… 385 58
## 5 1_AAGTCGT… AAGTCG… 1 BCB1… -2.30e-2 -0.0581 G1 MAIT 352 42
## 6 1_AATGAAG… AATGAA… 1 BCB1… 1.09e-2 -0.0621 S CD4+_r… 335 40
## 7 1_ACAAAGA… ACAAAG… 1 BCB1… -2.06e-2 -0.0409 G1 CD4+_T… 242 39
## 8 1_ACACGCG… ACACGC… 1 BCB1… -3.95e-4 -0.176 G1 CD8+_h… 438 45
## 9 1_ACATGCA… ACATGC… 1 BCB1… -4.09e-2 -0.0829 G1 CD4+_r… 180 39
## 10 1_ACGATCA… ACGATC… 1 BCB1… 8.82e-2 -0.0397 S CD4+_r… 82 34
## # … with 3,570 more rows, 8 more variables: nCount_SCT <int>,
## # nFeature_SCT <int>, ident <fct>, file <chr>, treatment <chr>,
## # sample <glue>, UMAP_1 <dbl>, UMAP_2 <dbl>, and abbreviated variable names
## # ¹G2M.Score, ²cell_type, ³nCount_RNA, ⁴nFeature_RNA
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesIf we want to revert to the standard SingleCellExperiment view we can do that.
options("restore_SingleCellExperiment_show" = TRUE)
sce_obj## class: SingleCellExperiment
## dim: 482 3580
## metadata(0):
## assays(2): counts logcounts
## rownames(482): HBB HBA2 ... TRGC2 CD8A
## rowData names(0):
## colnames(3580): 1_AAATGGACAAGTTCGT-1 1_AACAAGAGTGTTGAGG-1 ...
## 10_TTTGTTGGTACACGTT-1 10_TTTGTTGGTGATAGAT-1
## colData names(15): Barcode batch ... treatment sampleIf we want to revert back to tidy SingleCellExperiment view we can.
options("restore_SingleCellExperiment_show" = FALSE)
sce_obj## # A SingleCellExperiment-tibble abstraction: 3,580 × 18
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell Barcode batch BCB S.Score G2M.S…¹ Phase cell_…² nCoun…³ nFeat…⁴
## <chr> <fct> <fct> <fct> <dbl> <dbl> <fct> <fct> <int> <int>
## 1 1_AAATGGA… AAATGG… 1 BCB1… -2.07e-2 -0.0602 G1 TCR_V_… 215 36
## 2 1_AACAAGA… AACAAG… 1 BCB1… 2.09e-2 -0.0357 S CD8+_t… 145 41
## 3 1_AACGTCA… AACGTC… 1 BCB1… -2.54e-2 -0.133 G1 CD8+_h… 356 44
## 4 1_AACTTCT… AACTTC… 1 BCB1… 2.85e-2 -0.163 S CD8+_t… 385 58
## 5 1_AAGTCGT… AAGTCG… 1 BCB1… -2.30e-2 -0.0581 G1 MAIT 352 42
## 6 1_AATGAAG… AATGAA… 1 BCB1… 1.09e-2 -0.0621 S CD4+_r… 335 40
## 7 1_ACAAAGA… ACAAAG… 1 BCB1… -2.06e-2 -0.0409 G1 CD4+_T… 242 39
## 8 1_ACACGCG… ACACGC… 1 BCB1… -3.95e-4 -0.176 G1 CD8+_h… 438 45
## 9 1_ACATGCA… ACATGC… 1 BCB1… -4.09e-2 -0.0829 G1 CD4+_r… 180 39
## 10 1_ACGATCA… ACGATC… 1 BCB1… 8.82e-2 -0.0397 S CD4+_r… 82 34
## # … with 3,570 more rows, 8 more variables: nCount_SCT <int>,
## # nFeature_SCT <int>, ident <fct>, file <chr>, treatment <chr>,
## # sample <glue>, UMAP_1 <dbl>, UMAP_2 <dbl>, and abbreviated variable names
## # ¹G2M.Score, ²cell_type, ³nCount_RNA, ⁴nFeature_RNA
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesIt can be interacted with using SingleCellExperiment
commands such as assays.
assays(sce_obj)## List of length 2
## names(2): counts logcountsWe can also interact with our object as we do with any tidyverse tibble.
Tidyverse commands
We can use tidyverse commands, such as filter,
select and mutate to explore the
tidySingleCellExperiment object. Some examples are shown below and more
can be seen at the tidySingleCellExperiment website here.
We can use filter to choose rows, for example, to see
just the rows for the cells in G1 cell-cycle stage.
sce_obj |> filter(Phase == "G1")## # A SingleCellExperiment-tibble abstraction: 1,859 × 18
## # Features=482 | Cells=1859 | Assays=counts, logcounts
## .cell Barcode batch BCB S.Score G2M.S…¹ Phase cell_…² nCoun…³ nFeat…⁴
## <chr> <fct> <fct> <fct> <dbl> <dbl> <fct> <fct> <int> <int>
## 1 1_AAATGGA… AAATGG… 1 BCB1… -2.07e-2 -0.0602 G1 TCR_V_… 215 36
## 2 1_AACGTCA… AACGTC… 1 BCB1… -2.54e-2 -0.133 G1 CD8+_h… 356 44
## 3 1_AAGTCGT… AAGTCG… 1 BCB1… -2.30e-2 -0.0581 G1 MAIT 352 42
## 4 1_ACAAAGA… ACAAAG… 1 BCB1… -2.06e-2 -0.0409 G1 CD4+_T… 242 39
## 5 1_ACACGCG… ACACGC… 1 BCB1… -3.95e-4 -0.176 G1 CD8+_h… 438 45
## 6 1_ACATGCA… ACATGC… 1 BCB1… -4.09e-2 -0.0829 G1 CD4+_r… 180 39
## 7 1_ACGTAAC… ACGTAA… 1 BCB1… -8.69e-3 -0.140 G1 TCR_V_… 187 57
## 8 1_AGAGAAT… AGAGAA… 1 BCB1… -5.14e-3 -0.102 G1 CD8+_t… 155 40
## 9 1_AGATCCA… AGATCC… 1 BCB1… -1.17e-2 -0.155 G1 CD8+_h… 433 42
## 10 1_AGCCAAT… AGCCAA… 1 BCB1… -3.61e-2 -0.207 G1 CD4+_T… 353 43
## # … with 1,849 more rows, 8 more variables: nCount_SCT <int>,
## # nFeature_SCT <int>, ident <fct>, file <chr>, treatment <chr>,
## # sample <glue>, UMAP_1 <dbl>, UMAP_2 <dbl>, and abbreviated variable names
## # ¹G2M.Score, ²cell_type, ³nCount_RNA, ⁴nFeature_RNA
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesWe can use select to view columns, for example, to see
the filename, total cellular RNA abundance and cell phase.
- If we use
selectwe will also get any view-only columns returned, such as the UMAP columns generated during the preprocessing.
sce_obj |> select(.cell, file, nCount_RNA, Phase)## # A SingleCellExperiment-tibble abstraction: 3,580 × 6
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell file nCoun…¹ Phase UMAP_1 UMAP_2
## <chr> <chr> <int> <fct> <dbl> <dbl>
## 1 1_AAATGGACAAGTTCGT-1 ../data/single_cell/SI-GA-H… 215 G1 -3.73 -1.59
## 2 1_AACAAGAGTGTTGAGG-1 ../data/single_cell/SI-GA-H… 145 S 0.798 -0.151
## 3 1_AACGTCAGTCTATGAC-1 ../data/single_cell/SI-GA-H… 356 G1 -0.292 0.515
## 4 1_AACTTCTCACGCTGAC-1 ../data/single_cell/SI-GA-H… 385 S 0.372 2.34
## 5 1_AAGTCGTGTGTTGCCG-1 ../data/single_cell/SI-GA-H… 352 G1 -1.63 -0.236
## 6 1_AATGAAGCATCCAACA-1 ../data/single_cell/SI-GA-H… 335 S 0.822 2.90
## 7 1_ACAAAGAGTCGTACTA-1 ../data/single_cell/SI-GA-H… 242 G1 3.28 3.97
## 8 1_ACACGCGCAGGTACGA-1 ../data/single_cell/SI-GA-H… 438 G1 -3.65 -0.192
## 9 1_ACATGCATCACTTTGT-1 ../data/single_cell/SI-GA-H… 180 G1 -0.273 4.09
## 10 1_ACGATCATCGACGAGA-1 ../data/single_cell/SI-GA-H… 82 S -0.816 2.90
## # … with 3,570 more rows, and abbreviated variable name ¹nCount_RNA
## # ℹ Use `print(n = ...)` to see more rowsWe can use mutate to create a column. For example, we
could create a new Phase_l column that contains a
lower-case version of Phase.
## # A SingleCellExperiment-tibble abstraction: 3,580 × 5
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell Phase Phase_l UMAP_1 UMAP_2
## <chr> <fct> <chr> <dbl> <dbl>
## 1 1_AAATGGACAAGTTCGT-1 G1 g1 -3.73 -1.59
## 2 1_AACAAGAGTGTTGAGG-1 S s 0.798 -0.151
## 3 1_AACGTCAGTCTATGAC-1 G1 g1 -0.292 0.515
## 4 1_AACTTCTCACGCTGAC-1 S s 0.372 2.34
## 5 1_AAGTCGTGTGTTGCCG-1 G1 g1 -1.63 -0.236
## 6 1_AATGAAGCATCCAACA-1 S s 0.822 2.90
## 7 1_ACAAAGAGTCGTACTA-1 G1 g1 3.28 3.97
## 8 1_ACACGCGCAGGTACGA-1 G1 g1 -3.65 -0.192
## 9 1_ACATGCATCACTTTGT-1 G1 g1 -0.273 4.09
## 10 1_ACGATCATCGACGAGA-1 S s -0.816 2.90
## # … with 3,570 more rows
## # ℹ Use `print(n = ...)` to see more rowsWe can use tidyverse commands to polish an annotation column. We will extract the sample, and group information from the file name column into separate columns.
# First take a look at the file column
sce_obj |> select(.cell, file)## # A SingleCellExperiment-tibble abstraction: 3,580 × 4
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell file UMAP_1 UMAP_2
## <chr> <chr> <dbl> <dbl>
## 1 1_AAATGGACAAGTTCGT-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -3.73 -1.59
## 2 1_AACAAGAGTGTTGAGG-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… 0.798 -0.151
## 3 1_AACGTCAGTCTATGAC-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -0.292 0.515
## 4 1_AACTTCTCACGCTGAC-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… 0.372 2.34
## 5 1_AAGTCGTGTGTTGCCG-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -1.63 -0.236
## 6 1_AATGAAGCATCCAACA-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… 0.822 2.90
## 7 1_ACAAAGAGTCGTACTA-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… 3.28 3.97
## 8 1_ACACGCGCAGGTACGA-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -3.65 -0.192
## 9 1_ACATGCATCACTTTGT-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -0.273 4.09
## 10 1_ACGATCATCGACGAGA-1 ../data/single_cell/SI-GA-H1/outs/raw_fea… -0.816 2.90
## # … with 3,570 more rows
## # ℹ Use `print(n = ...)` to see more rows
# Create column for sample
sce_obj <- sce_obj |>
# Extract sample
extract(file, "sample", "../data/.*/([a-zA-Z0-9_-]+)/outs.+", remove = FALSE)
# Take a look
sce_obj |> select(.cell, sample, everything())## # A SingleCellExperiment-tibble abstraction: 3,580 × 18
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell sample Barcode batch BCB S.Score G2M.S…¹ Phase cell_…² nCoun…³
## <chr> <chr> <fct> <fct> <fct> <dbl> <dbl> <fct> <fct> <int>
## 1 1_AAATGGAC… SI-GA… AAATGG… 1 BCB1… -2.07e-2 -0.0602 G1 TCR_V_… 215
## 2 1_AACAAGAG… SI-GA… AACAAG… 1 BCB1… 2.09e-2 -0.0357 S CD8+_t… 145
## 3 1_AACGTCAG… SI-GA… AACGTC… 1 BCB1… -2.54e-2 -0.133 G1 CD8+_h… 356
## 4 1_AACTTCTC… SI-GA… AACTTC… 1 BCB1… 2.85e-2 -0.163 S CD8+_t… 385
## 5 1_AAGTCGTG… SI-GA… AAGTCG… 1 BCB1… -2.30e-2 -0.0581 G1 MAIT 352
## 6 1_AATGAAGC… SI-GA… AATGAA… 1 BCB1… 1.09e-2 -0.0621 S CD4+_r… 335
## 7 1_ACAAAGAG… SI-GA… ACAAAG… 1 BCB1… -2.06e-2 -0.0409 G1 CD4+_T… 242
## 8 1_ACACGCGC… SI-GA… ACACGC… 1 BCB1… -3.95e-4 -0.176 G1 CD8+_h… 438
## 9 1_ACATGCAT… SI-GA… ACATGC… 1 BCB1… -4.09e-2 -0.0829 G1 CD4+_r… 180
## 10 1_ACGATCAT… SI-GA… ACGATC… 1 BCB1… 8.82e-2 -0.0397 S CD4+_r… 82
## # … with 3,570 more rows, 8 more variables: nFeature_RNA <int>,
## # nCount_SCT <int>, nFeature_SCT <int>, ident <fct>, file <chr>,
## # treatment <chr>, UMAP_1 <dbl>, UMAP_2 <dbl>, and abbreviated variable names
## # ¹G2M.Score, ²cell_type, ³nCount_RNA
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesWe could use tidyverse unite to combine columns, for
example to create a new column for sample id combining the sample and
patient id (BCB) columns.
sce_obj <- sce_obj |> unite("sample_id", sample, BCB, remove = FALSE)
# Take a look
sce_obj |> select(.cell, sample_id, sample, BCB)## # A SingleCellExperiment-tibble abstraction: 3,580 × 6
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell sample_id sample BCB UMAP_1 UMAP_2
## <chr> <chr> <chr> <fct> <dbl> <dbl>
## 1 1_AAATGGACAAGTTCGT-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.73 -1.59
## 2 1_AACAAGAGTGTTGAGG-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 0.798 -0.151
## 3 1_AACGTCAGTCTATGAC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -0.292 0.515
## 4 1_AACTTCTCACGCTGAC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 0.372 2.34
## 5 1_AAGTCGTGTGTTGCCG-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -1.63 -0.236
## 6 1_AATGAAGCATCCAACA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 0.822 2.90
## 7 1_ACAAAGAGTCGTACTA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 3.28 3.97
## 8 1_ACACGCGCAGGTACGA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.65 -0.192
## 9 1_ACATGCATCACTTTGT-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -0.273 4.09
## 10 1_ACGATCATCGACGAGA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -0.816 2.90
## # … with 3,570 more rows
## # ℹ Use `print(n = ...)` to see more rowsPart 2 Signature visualisation
Data pre-processing
The object sce_obj we’ve been using was created as part
of a study on breast cancer systemic immune response. Peripheral blood
mononuclear cells have been sequenced for RNA at the single-cell level.
The steps used to generate the object are summarised below.
scran,scater, andDropletsUtilspackages have been used to eliminate empty droplets and dead cells. Samples were individually quality checked and cells were filtered for good gene coverage.Variable features were identified using
modelGeneVar.Read counts were scaled and normalised using logNormCounts from
scuttle.Data integration was performed using
fastMNNwith default parameters.PCA performed to reduce feature dimensionality.
Nearest-neighbor cell networks were calculated using 30 principal components.
2 UMAP dimensions were calculated using 30 principal components.
Cells with similar transcriptome profiles were grouped into clusters using Louvain clustering from
scran.
Analyse custom signature
The researcher analysing this dataset wanted to identify gamma delta T cells using a gene signature from a published paper (Pizzolato et al. 2019). We’ll show how that can be done here.
With tidySingleCellExperiment’s join_features we can
view the counts for genes in the signature as columns joined to our
single cell tibble.
sce_obj |>
join_features(c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide")## # A SingleCellExperiment-tibble abstraction: 3,580 × 25
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell Barcode batch sampl…¹ BCB S.Score G2M.S…² Phase cell_…³ nCoun…⁴
## <chr> <fct> <fct> <chr> <fct> <dbl> <dbl> <fct> <fct> <int>
## 1 1_AAATGGA… AAATGG… 1 SI-GA-… BCB1… -2.07e-2 -0.0602 G1 TCR_V_… 215
## 2 1_AACAAGA… AACAAG… 1 SI-GA-… BCB1… 2.09e-2 -0.0357 S CD8+_t… 145
## 3 1_AACGTCA… AACGTC… 1 SI-GA-… BCB1… -2.54e-2 -0.133 G1 CD8+_h… 356
## 4 1_AACTTCT… AACTTC… 1 SI-GA-… BCB1… 2.85e-2 -0.163 S CD8+_t… 385
## 5 1_AAGTCGT… AAGTCG… 1 SI-GA-… BCB1… -2.30e-2 -0.0581 G1 MAIT 352
## 6 1_AATGAAG… AATGAA… 1 SI-GA-… BCB1… 1.09e-2 -0.0621 S CD4+_r… 335
## 7 1_ACAAAGA… ACAAAG… 1 SI-GA-… BCB1… -2.06e-2 -0.0409 G1 CD4+_T… 242
## 8 1_ACACGCG… ACACGC… 1 SI-GA-… BCB1… -3.95e-4 -0.176 G1 CD8+_h… 438
## 9 1_ACATGCA… ACATGC… 1 SI-GA-… BCB1… -4.09e-2 -0.0829 G1 CD4+_r… 180
## 10 1_ACGATCA… ACGATC… 1 SI-GA-… BCB1… 8.82e-2 -0.0397 S CD4+_r… 82
## # … with 3,570 more rows, 15 more variables: nFeature_RNA <int>,
## # nCount_SCT <int>, nFeature_SCT <int>, ident <fct>, file <chr>,
## # sample <chr>, treatment <chr>, CD3D <dbl>, TRDC <dbl>, TRGC1 <dbl>,
## # TRGC2 <dbl>, CD8A <dbl>, CD8B <dbl>, UMAP_1 <dbl>, UMAP_2 <dbl>, and
## # abbreviated variable names ¹sample_id, ²G2M.Score, ³cell_type, ⁴nCount_RNA
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesWe can use tidyverse mutate to create a column
containing the signature score. To generate the score, we scale the sum
of the 4 genes, CD3D, TRDC, TRGC1, TRGC2, and subtract the scaled sum of
the 2 genes, CD8A and CD8B. mutate is powerful in enabling
us to perform complex arithmetic operations easily.
sce_obj |>
join_features(c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide") |>
mutate(
signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to = c(0, 1)) -
scales::rescale(CD8A + CD8B, to = c(0, 1))
) |>
select(.cell, signature_score, everything())## # A SingleCellExperiment-tibble abstraction: 3,580 × 26
## # Features=482 | Cells=3580 | Assays=counts, logcounts
## .cell signa…¹ Barcode batch sampl…² BCB S.Score G2M.S…³ Phase cell_…⁴
## <chr> <dbl> <fct> <fct> <chr> <fct> <dbl> <dbl> <fct> <fct>
## 1 1_AAATGGA… 0.386 AAATGG… 1 SI-GA-… BCB1… -2.07e-2 -0.0602 G1 TCR_V_…
## 2 1_AACAAGA… 0.108 AACAAG… 1 SI-GA-… BCB1… 2.09e-2 -0.0357 S CD8+_t…
## 3 1_AACGTCA… -0.789 AACGTC… 1 SI-GA-… BCB1… -2.54e-2 -0.133 G1 CD8+_h…
## 4 1_AACTTCT… -0.0694 AACTTC… 1 SI-GA-… BCB1… 2.85e-2 -0.163 S CD8+_t…
## 5 1_AAGTCGT… 0 AAGTCG… 1 SI-GA-… BCB1… -2.30e-2 -0.0581 G1 MAIT
## 6 1_AATGAAG… 0 AATGAA… 1 SI-GA-… BCB1… 1.09e-2 -0.0621 S CD4+_r…
## 7 1_ACAAAGA… 0.108 ACAAAG… 1 SI-GA-… BCB1… -2.06e-2 -0.0409 G1 CD4+_T…
## 8 1_ACACGCG… 0.170 ACACGC… 1 SI-GA-… BCB1… -3.95e-4 -0.176 G1 CD8+_h…
## 9 1_ACATGCA… 0 ACATGC… 1 SI-GA-… BCB1… -4.09e-2 -0.0829 G1 CD4+_r…
## 10 1_ACGATCA… 0.278 ACGATC… 1 SI-GA-… BCB1… 8.82e-2 -0.0397 S CD4+_r…
## # … with 3,570 more rows, 16 more variables: nCount_RNA <int>,
## # nFeature_RNA <int>, nCount_SCT <int>, nFeature_SCT <int>, ident <fct>,
## # file <chr>, sample <chr>, treatment <chr>, CD3D <dbl>, TRDC <dbl>,
## # TRGC1 <dbl>, TRGC2 <dbl>, CD8A <dbl>, CD8B <dbl>, UMAP_1 <dbl>,
## # UMAP_2 <dbl>, and abbreviated variable names ¹signature_score, ²sample_id,
## # ³G2M.Score, ⁴cell_type
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesThe gamma delta T cells could then be visualised by the signature score using Bioconductor’s visualisation functions.
sce_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide"
) |>
mutate(
signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to = c(0, 1)) -
scales::rescale(CD8A + CD8B, to = c(0, 1))
) |>
scater::plotUMAP(colour_by = "signature_score")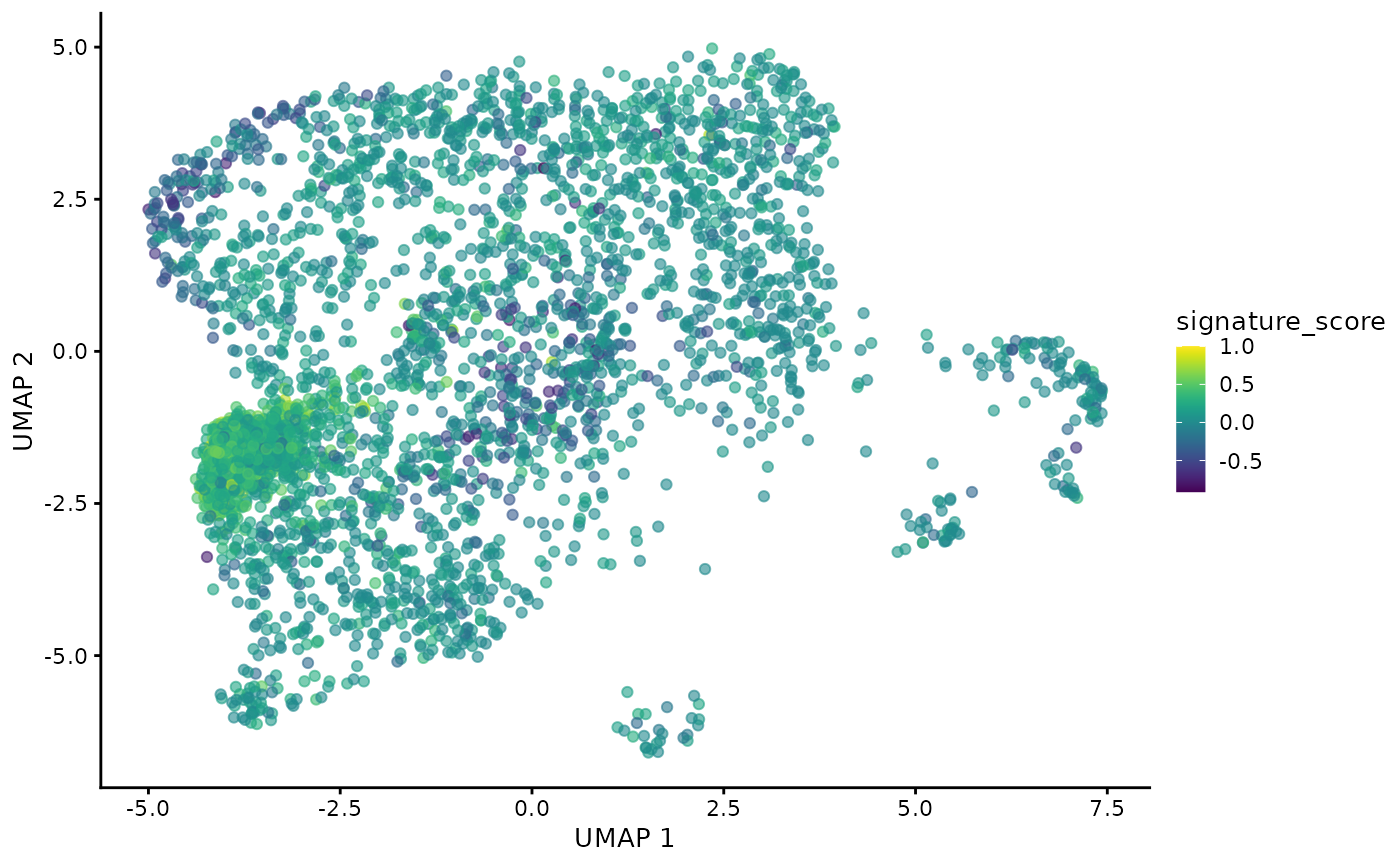
The cells could also be visualised using the popular and powerful
ggplot2 package, enabling the researcher to use ggplot
functions they were familiar with, and to customise the plot with great
flexibility.
sce_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide"
) |>
mutate(
signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to = c(0, 1)) -
scales::rescale(CD8A + CD8B, to = c(0, 1))
) |>
# plot cells with high score last so they're not obscured by other cells
arrange(signature_score) |>
ggplot(aes(UMAP_1, UMAP_2, color = signature_score)) +
geom_point() +
scale_color_distiller(palette = "Spectral") +
bioc2022tidytranscriptomics::theme_multipanel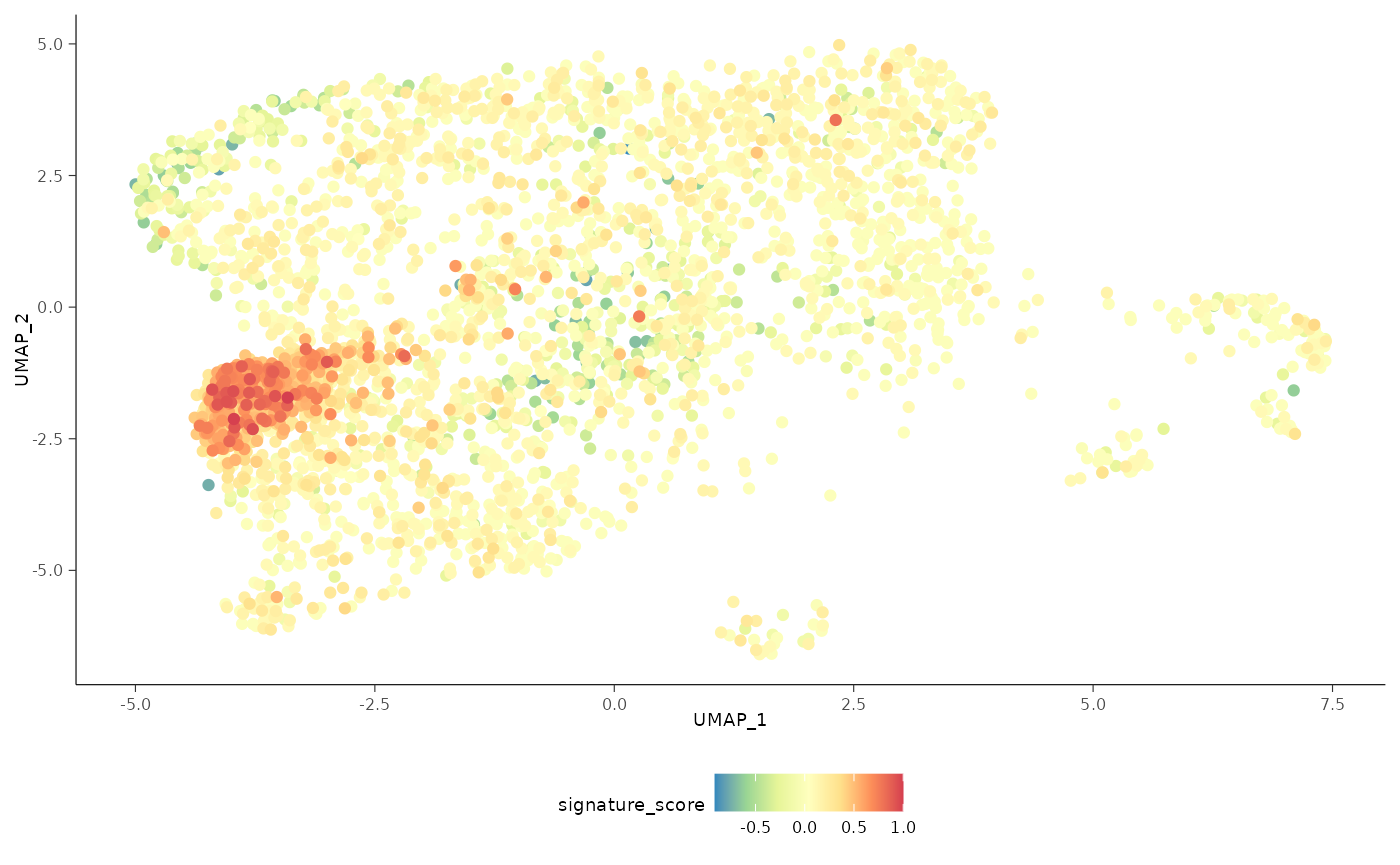
For exploratory analyses, we can select the gamma delta T cells, the red cluster on the left with high signature score. We’ll filter for cells with a signature score > 0.7.
sce_obj_gamma_delta <-
sce_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide"
) |>
mutate(
signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to = c(0, 1)) -
scales::rescale(CD8A + CD8B, to = c(0, 1))
) |>
# Proper cluster selection should be used instead (see supplementary material)
filter(signature_score > 0.7)For comparison, we show the alternative using base R and SingleCellExperiment. Note that the code contains more redundancy and intermediate objects.
counts_positive <-
assay(sce_obj, "logcounts")[c("CD3D", "TRDC", "TRGC1", "TRGC2"), ] |>
colSums() |>
scales::rescale(to = c(0, 1))
counts_negative <-
assay(sce_obj, "logcounts")[c("CD8A", "CD8B"), ] |>
colSums() |>
scales::rescale(to = c(0, 1))
sce_obj$signature_score <- counts_positive - counts_negative
sce_obj_gamma_delta <- sce_obj[, sce_obj$signature_score > 0.7]We can then focus on just these gamma delta T cells and chain Bioconductor and tidyverse commands together to analyse.
library(batchelor)
library(scater)
sce_obj_gamma_delta <-
sce_obj_gamma_delta |>
# Integrate - using batchelor.
multiBatchNorm(batch = colData(sce_obj_gamma_delta)$sample) |>
fastMNN(batch = colData(sce_obj_gamma_delta)$sample) |>
# Join metadata removed by fastMNN - using tidyverse
left_join(as_tibble(sce_obj_gamma_delta)) |>
# Dimension reduction - using scater
runUMAP(ncomponents = 2, dimred = "corrected")Visualise gamma delta T cells. As we have used rough threshold we are left with only few cells. Proper cluster selection should be used instead (see supplementary material).
sce_obj_gamma_delta |> plotUMAP() 
It is also possible to visualise the cells as a 3D plot using plotly. The example data used here only contains a few genes, for the sake of time and size in this demonstration, but below is how you could generate the 3 dimensions needed for 3D plot with a full dataset.
single_cell_object |>
RunUMAP(dims = 1:30, n.components = 3L, spread = 0.5, min.dist = 0.01, n.neighbors = 10L)We’ll demonstrate creating a 3D plot using some data that has 3 UMAP dimensions. This is a fantastic way to visualise both reduced dimensions and metadata in the same representation.
pbmc <- bioc2022tidytranscriptomics::sce_obj_UMAP3
pbmc |>
plot_ly(
x = ~`UMAP_1`,
y = ~`UMAP_2`,
z = ~`UMAP_3`,
color = ~cell_type,
colors = dittoSeq::dittoColors()
) %>%
add_markers(size = I(1))Part 3 Pseudobulk analyses
Next we want to identify genes whose transcription is affected by treatment in this dataset, comparing treated and untreated patients. We can do this with pseudobulk analysis. We aggregate cell-wise transcript abundance into pseudobulk samples and can then perform hypothesis testing using the very well established bulk RNA sequencing tools. For example, we can use edgeR in tidybulk to perform differential expression testing. For more details on pseudobulk analysis see here.
We want to do it for each cell type and the tidy transcriptomics ecosystem makes this very easy.
Create pseudobulk samples
To create pseudobulk samples from the single cell samples, we will
use a helper function called aggregate_cells, available in
this workshop package. This function will combine the single cells into
a group for each cell type for each sample.
pseudo_bulk <-
sce_obj |>
bioc2022tidytranscriptomics::aggregate_cells(c(sample, cell_type), assays = "counts")
pseudo_bulk## # A SummarizedExperiment-tibble abstraction: 55,430 × 115
## # Features=482 | Samples=115 | Assays=counts
## .feature .sample counts sample cell_…¹ .aggr…² batch sampl…³ BCB file
## <chr> <chr> <dbl> <chr> <fct> <int> <fct> <chr> <fct> <chr>
## 1 ABCC3 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 2 AC004585.1 SI-GA-E5_… 1 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 3 AC005480.1 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 4 AC007952.4 SI-GA-E5_… 1 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 5 AC012615.2 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 6 AC020656.1 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 7 AC021739.4 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 8 AC026979.2 SI-GA-E5_… 1 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 9 AC046158.1 SI-GA-E5_… 0 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## 10 AC055713.1 SI-GA-E5_… 1 SI-GA… CD4+_r… 24 2 SI-GA-… BCB0… ../d…
## # … with 40 more rows, 2 more variables: treatment <chr>, feature <chr>, and
## # abbreviated variable names ¹cell_type, ².aggregated_cells, ³sample_id
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesTidybulk and tidySummarizedExperiment
With tidySummarizedExperiment and tidybulk
it is easy to split the data into groups and perform analyses on each
without needing to create separate objects.
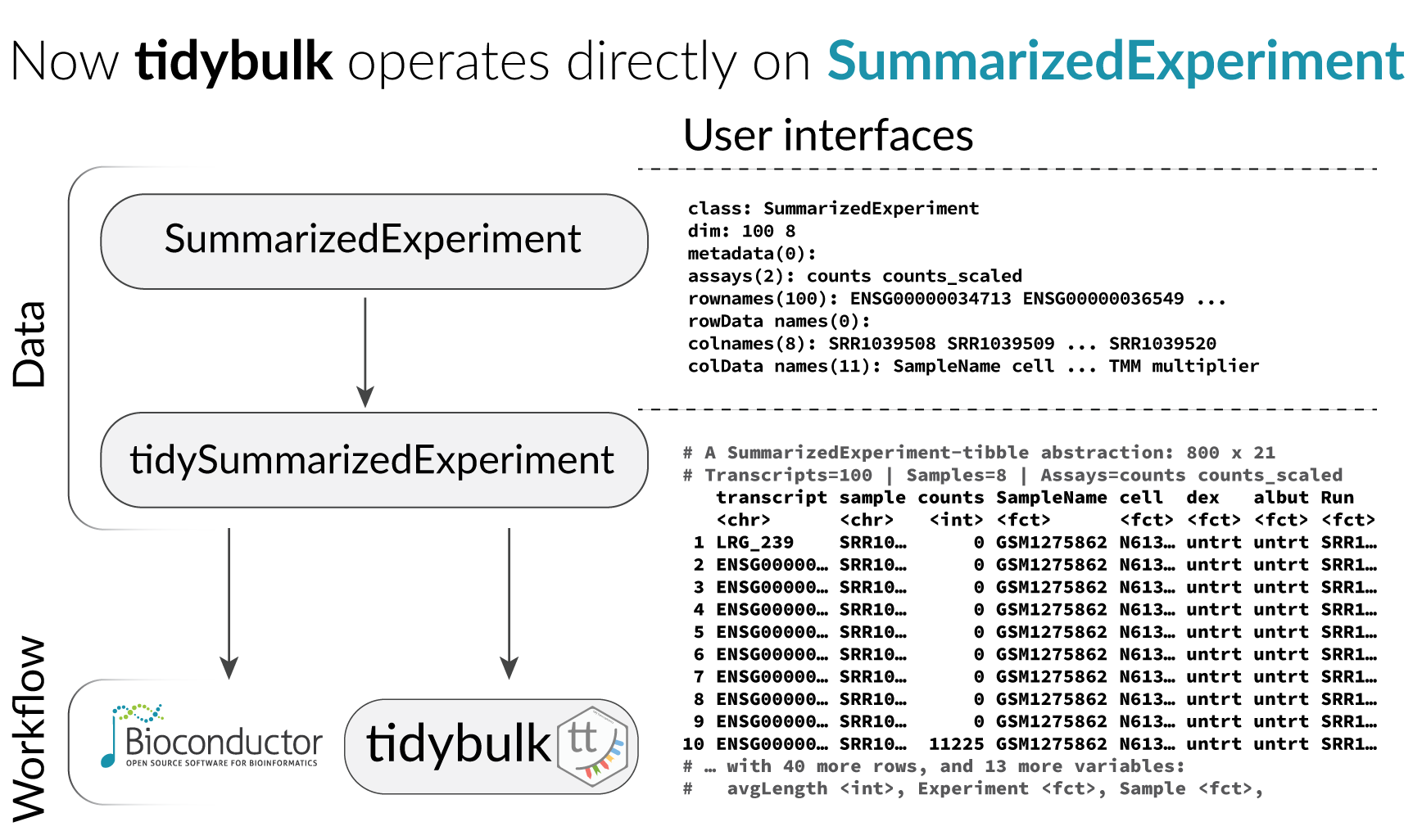
We use tidyverse nest to group the data. The command
below will create a tibble containing a column with a
SummarizedExperiment object for each cell type. nest is
similar to tidyverse group_by, except with
nest each group is stored in a single row, and can be a
complex object such as a plot or SummarizedExperiment.
pseudo_bulk_nested <-
pseudo_bulk |>
nest(grouped_summarized_experiment = -cell_type)
pseudo_bulk_nested## # A tibble: 13 × 2
## cell_type grouped_summarized_experiment
## <fct> <list>
## 1 CD4+_ribosome_rich <SmmrzdEx[,10]>
## 2 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]>
## 3 CD8+_high_ribonucleosome <SmmrzdEx[,10]>
## 4 CD8+_Tcm <SmmrzdEx[,6]>
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <SmmrzdEx[,10]>
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]>
## 7 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,3]>
## 8 MAIT <SmmrzdEx[,10]>
## 9 NK_cells <SmmrzdEx[,10]>
## 10 NK_cycling <SmmrzdEx[,7]>
## 11 T_cell:CD8+_other <SmmrzdEx[,10]>
## 12 TCR_V_Delta_1 <SmmrzdEx[,10]>
## 13 TCR_V_Delta_2 <SmmrzdEx[,9]>To explore the grouping, we can use tidyverse slice to
choose a row (cell_type) and pull to extract the values
from a column. If we pull the data column we can view the
SummarizedExperiment object.
## [[1]]
## # A SummarizedExperiment-tibble abstraction: 4,820 × 10
## # Features=482 | Samples=10 | Assays=counts
## .feature .sample counts sample .aggr…¹ batch sampl…² BCB file treat…³
## <chr> <chr> <dbl> <chr> <int> <fct> <chr> <fct> <chr> <chr>
## 1 ABCC3 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 2 AC004585.1 SI-GA-E5_… 1 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 3 AC005480.1 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 4 AC007952.4 SI-GA-E5_… 1 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 5 AC012615.2 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 6 AC020656.1 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 7 AC021739.4 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 8 AC026979.2 SI-GA-E5_… 1 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 9 AC046158.1 SI-GA-E5_… 0 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## 10 AC055713.1 SI-GA-E5_… 1 SI-GA… 24 2 SI-GA-… BCB0… ../d… treated
## # … with 40 more rows, 1 more variable: feature <chr>, and abbreviated variable
## # names ¹.aggregated_cells, ²sample_id, ³treatment
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesWe can then identify differentially expressed genes for each cell
type for our condition of interest, treated versus untreated patients.
We use tidyverse map to apply differential expression
functions to each cell type group in the nested data. The result columns
will be added to the SummarizedExperiment objects.
# Differential transcription abundance
pseudo_bulk <-
pseudo_bulk_nested |>
# map accepts a data column (.x) and a function. It applies the function to each element of the column.
mutate(grouped_summarized_experiment = map(
grouped_summarized_experiment,
~ .x |>
# Removing genes with low expression
identify_abundant(factor_of_interest = treatment) |>
# Scaling counts for sequencing depth
scale_abundance(method="TMMwsp") |>
# Testing for differential expression using edgeR quasi likelihood
test_differential_abundance(~treatment, method="edgeR_quasi_likelihood", scaling_method="TMMwsp")
))The output is again a tibble containing a SummarizedExperiment object for each cell type.
pseudo_bulk## # A tibble: 13 × 2
## cell_type grouped_summarized_experiment
## <fct> <list>
## 1 CD4+_ribosome_rich <SmmrzdEx[,10]>
## 2 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]>
## 3 CD8+_high_ribonucleosome <SmmrzdEx[,10]>
## 4 CD8+_Tcm <SmmrzdEx[,6]>
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <SmmrzdEx[,10]>
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]>
## 7 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,3]>
## 8 MAIT <SmmrzdEx[,10]>
## 9 NK_cells <SmmrzdEx[,10]>
## 10 NK_cycling <SmmrzdEx[,7]>
## 11 T_cell:CD8+_other <SmmrzdEx[,10]>
## 12 TCR_V_Delta_1 <SmmrzdEx[,10]>
## 13 TCR_V_Delta_2 <SmmrzdEx[,9]>If we pull out the SummarizedExperiment object for the first cell type, as before, we can see it now has columns containing the differential expression results (e.g. logFC, PValue).
## [[1]]
## # A SummarizedExperiment-tibble abstraction: 4,820 × 10
## # Features=482 | Samples=10 | Assays=counts, counts_scaled
## .feature .sample counts count…¹ sample .aggr…² batch sampl…³ BCB file
## <chr> <chr> <dbl> <dbl> <chr> <int> <fct> <chr> <fct> <chr>
## 1 ABCC3 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 2 AC004585.1 SI-GA-E5_… 1 6.12 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 3 AC005480.1 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 4 AC007952.4 SI-GA-E5_… 1 6.12 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 5 AC012615.2 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 6 AC020656.1 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 7 AC021739.4 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 8 AC026979.2 SI-GA-E5_… 1 6.12 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 9 AC046158.1 SI-GA-E5_… 0 0 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## 10 AC055713.1 SI-GA-E5_… 1 6.12 SI-GA… 24 2 SI-GA-… BCB0… ../d…
## # … with 40 more rows, 10 more variables: treatment <chr>, TMM <dbl>,
## # multiplier <dbl>, feature <chr>, .abundant <lgl>, logFC <dbl>,
## # logCPM <dbl>, F <dbl>, PValue <dbl>, FDR <dbl>, and abbreviated variable
## # names ¹counts_scaled, ².aggregated_cells, ³sample_id
## # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesIt is useful to create plots for significant genes to visualise the transcriptional abundance in the groups being compared (treated and untreated). We can do this for each cell type without needing to create multiple objects.
pseudo_bulk <-
pseudo_bulk |>
# Filter out significant
# using a high FDR value as this is toy data
mutate(grouped_summarized_experiment = map(
grouped_summarized_experiment,
~ filter(.x, FDR < 0.5)
)) |>
# Filter cell types with no differential abundant gene-transcripts
# map_int is map that returns integer instead of list
filter(map_int(grouped_summarized_experiment, ~ nrow(.x)) > 0) |>
# Plot significant genes for each cell type
# map2 is map that accepts 2 input columns (.x, .y) and a function
mutate(plot = map2(
grouped_summarized_experiment, cell_type,
~ .x |>
ggplot(aes(treatment, counts_scaled + 1)) +
geom_boxplot(aes(fill = treatment)) +
geom_jitter() +
scale_y_log10() +
facet_wrap(~.feature) +
ggtitle(.y)
))The output is a nested table with a column containing a plot for each cell type.
pseudo_bulk## # A tibble: 5 × 3
## cell_type grouped_summarized_experiment plot
## <fct> <list> <list>
## 1 CD4+_ribosome_rich <SmmrzdEx[,10]> <gg>
## 2 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,3]> <gg>
## 3 NK_cells <SmmrzdEx[,10]> <gg>
## 4 T_cell:CD8+_other <SmmrzdEx[,10]> <gg>
## 5 TCR_V_Delta_1 <SmmrzdEx[,10]> <gg>We’ll use slice and pull again to have a look at one of the plots.
## [[1]]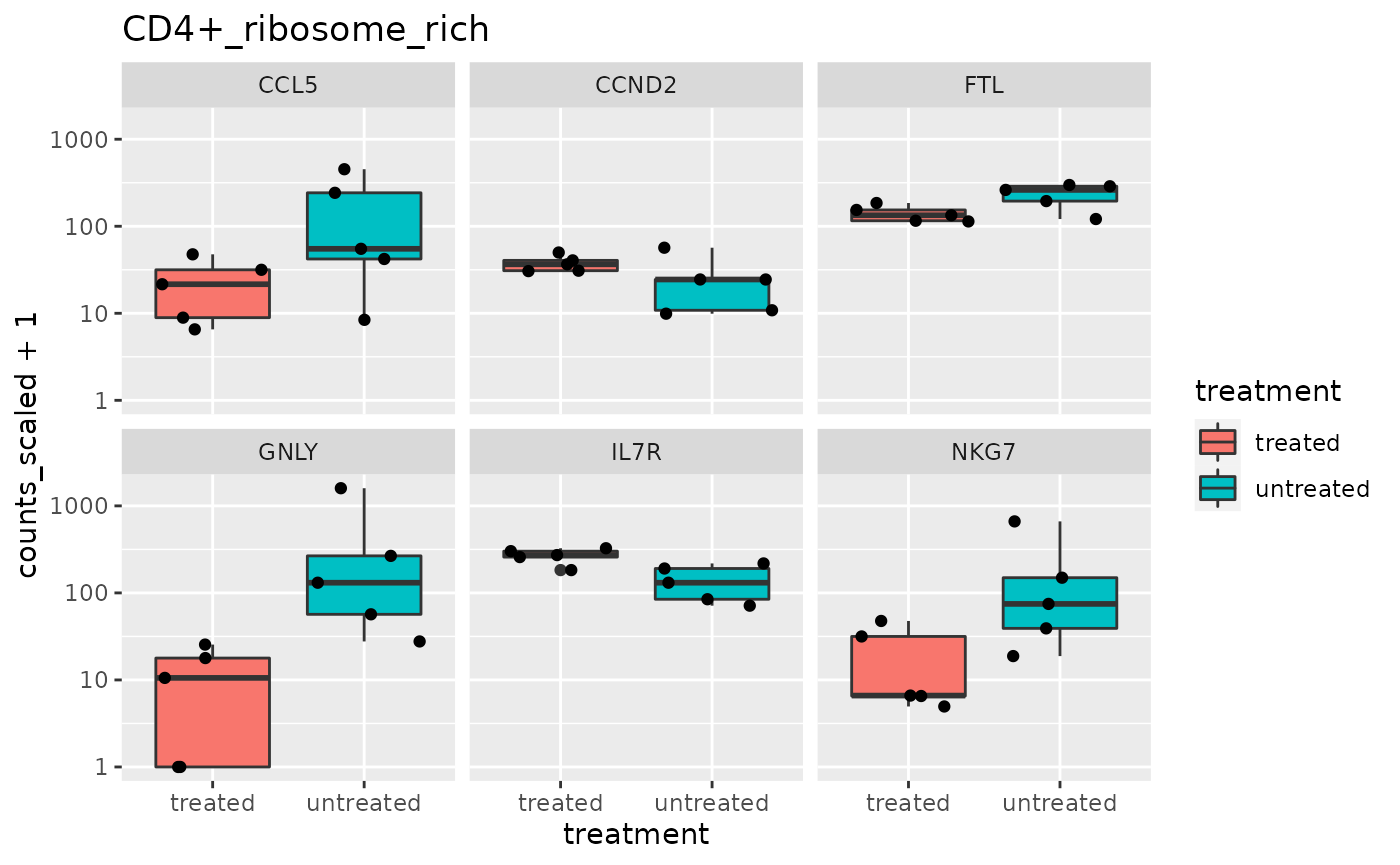
We can extract all plots and plot with wrap_plots from
the patchwork package.
pseudo_bulk |>
pull(plot) |>
wrap_plots() &
bioc2022tidytranscriptomics::theme_multipanel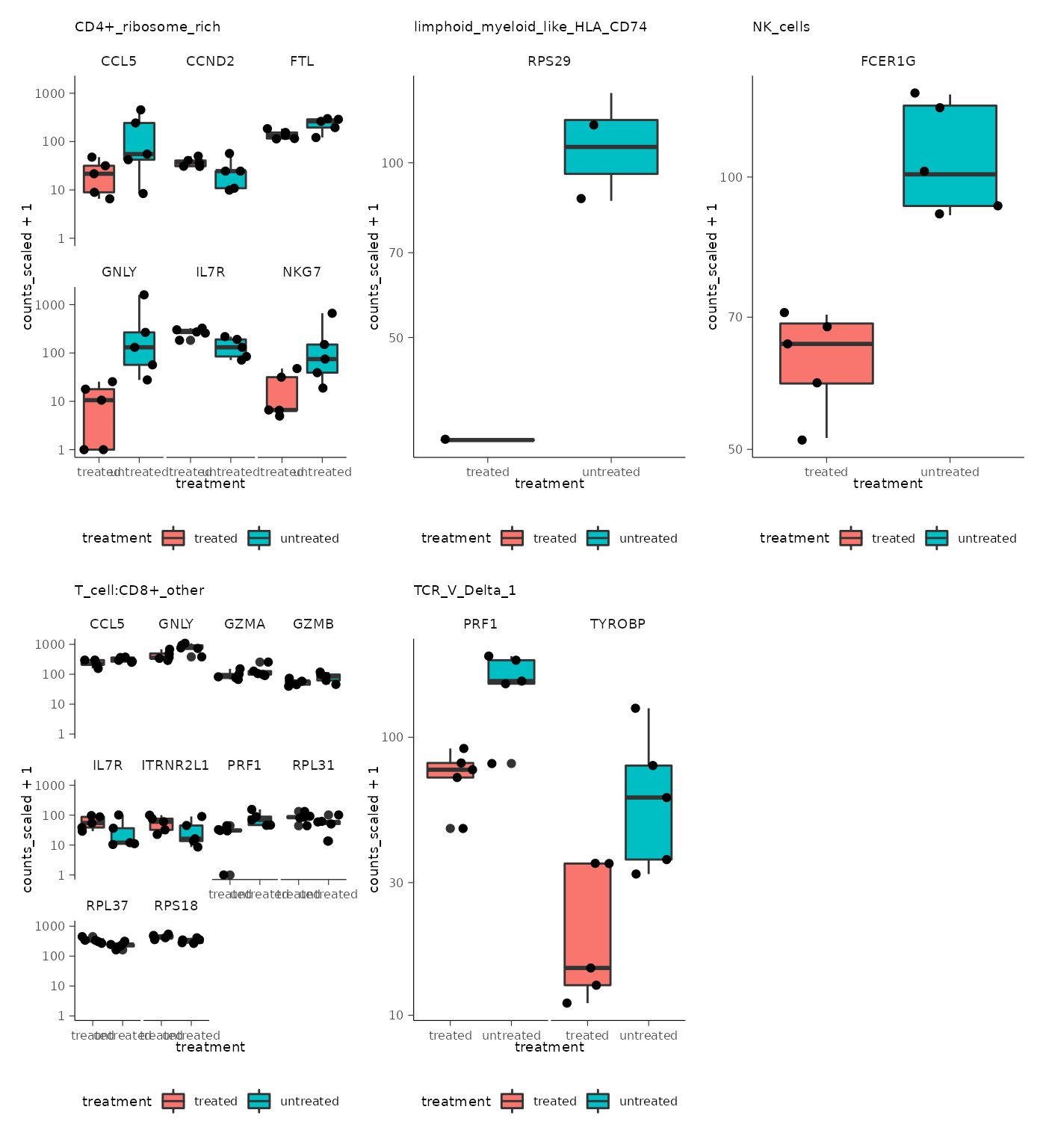
Feedback
Thank you for attending this workshop. We hope it was an informative session for you. We would be grateful if you could help us by taking a few moments to provide your valuable feedback in the short form below. Your feedback will provide us with an opportunity to further improve the workshop. All the results are anonymous.
Session Information
## R version 4.2.0 (2022-04-22)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] tidySummarizedExperiment_1.7.3 tidybulk_1.9.2
## [3] patchwork_1.1.1 purrr_0.3.4
## [5] tidyr_1.2.0 glue_1.6.2
## [7] scater_1.25.2 scuttle_1.7.2
## [9] batchelor_1.13.3 tidySingleCellExperiment_1.7.4
## [11] ttservice_0.2.2 dittoSeq_1.9.1
## [13] colorspace_2.0-3 dplyr_1.0.9
## [15] plotly_4.10.0 ggplot2_3.3.6
## [17] SingleCellExperiment_1.19.0 SummarizedExperiment_1.27.1
## [19] Biobase_2.57.1 GenomicRanges_1.49.0
## [21] GenomeInfoDb_1.33.3 IRanges_2.31.0
## [23] S4Vectors_0.35.1 BiocGenerics_0.43.1
## [25] MatrixGenerics_1.9.1 matrixStats_0.62.0
##
## loaded via a namespace (and not attached):
## [1] ggbeeswarm_0.6.0 ellipsis_0.3.2
## [3] bioc2022tidytranscriptomics_0.13.3 ggridges_0.5.3
## [5] rprojroot_2.0.3 XVector_0.37.0
## [7] BiocNeighbors_1.15.1 fs_1.5.2
## [9] farver_2.1.1 ggrepel_0.9.1
## [11] RSpectra_0.16-1 fansi_1.0.3
## [13] splines_4.2.0 codetools_0.2-18
## [15] sparseMatrixStats_1.9.0 cachem_1.0.6
## [17] knitr_1.39 jsonlite_1.8.0
## [19] ResidualMatrix_1.7.0 pheatmap_1.0.12
## [21] uwot_0.1.11 BiocManager_1.30.18
## [23] readr_2.1.2 compiler_4.2.0
## [25] httr_1.4.3 Matrix_1.4-1
## [27] fastmap_1.1.0 lazyeval_0.2.2
## [29] limma_3.53.5 cli_3.3.0
## [31] BiocSingular_1.13.0 htmltools_0.5.3
## [33] tools_4.2.0 rsvd_1.0.5
## [35] igraph_1.3.4 gtable_0.3.0
## [37] GenomeInfoDbData_1.2.8 Rcpp_1.0.9
## [39] jquerylib_0.1.4 pkgdown_2.0.6
## [41] vctrs_0.4.1 preprocessCore_1.59.0
## [43] crosstalk_1.2.0 DelayedMatrixStats_1.19.0
## [45] xfun_0.31 stringr_1.4.0
## [47] beachmat_2.13.4 lifecycle_1.0.1
## [49] irlba_2.3.5 edgeR_3.39.3
## [51] zlibbioc_1.43.0 scales_1.2.0
## [53] ragg_1.2.2 hms_1.1.1
## [55] parallel_4.2.0 RColorBrewer_1.1-3
## [57] yaml_2.3.5 memoise_2.0.1
## [59] gridExtra_2.3 sass_0.4.2
## [61] stringi_1.7.8 highr_0.9
## [63] desc_1.4.1 ScaledMatrix_1.5.0
## [65] BiocParallel_1.31.10 rlang_1.0.4
## [67] pkgconfig_2.0.3 systemfonts_1.0.4
## [69] bitops_1.0-7 evaluate_0.15
## [71] lattice_0.20-45 htmlwidgets_1.5.4
## [73] labeling_0.4.2 cowplot_1.1.1
## [75] tidyselect_1.1.2 plyr_1.8.7
## [77] magrittr_2.0.3 R6_2.5.1
## [79] generics_0.1.3 DelayedArray_0.23.0
## [81] pillar_1.8.0 withr_2.5.0
## [83] RCurl_1.98-1.7 tibble_3.1.8
## [85] crayon_1.5.1 utf8_1.2.2
## [87] tzdb_0.3.0 rmarkdown_2.14
## [89] viridis_0.6.2 locfit_1.5-9.5
## [91] grid_4.2.0 data.table_1.14.2
## [93] FNN_1.1.3.1 digest_0.6.29
## [95] textshaping_0.3.6 munsell_0.5.0
## [97] beeswarm_0.4.0 viridisLite_0.4.0
## [99] vipor_0.4.5 bslib_0.4.0References