Tidy Transcriptomics for Single-cell RNA Sequencing Analyses
Maria Doyle, Peter MacCallum Cancer Centre1
Stefano Mangiola, Walter and Eliza Hall Institute2
Source:vignettes/main.Rmd
main.RmdInstructors
Dr. Stefano Mangiola is currently a Postdoctoral researcher in the laboratory of Prof. Tony Papenfuss at the Walter and Eliza Hall Institute in Melbourne, Australia. His background spans from biotechnology to bioinformatics and biostatistics. His research focuses on prostate and breast tumour microenvironment, the development of statistical models for the analysis of RNA sequencing data, and data analysis and visualisation interfaces.
Workshop goals and objectives
What you will learn
- Basic
tidyoperations possible withtidyseuratandtidySingleCellExperiment - The differences between
SeuratandSingleCellExperimentrepresentation, andtidyrepresentation - How to interface
SeuratandSingleCellExperimentwith tidy manipulation and visualisation - A real-world case study that will showcase the power of
tidysingle-cell methods compared with base/ad-hoc methods
Part 1 Introduction to tidyseurat
# Load packages
library(purrr)
library(Seurat)
library(ggplot2)
library(dplyr)
library(colorspace)
library(dittoSeq)Seurat is a very popular analysis toolkit for single cell RNA sequencing data (Butler et al. 2018; Stuart et al. 2019).
Here we load single-cell data in Seurat object format. This data is peripheral blood mononuclear cells (PBMCs) from metastatic breast cancer patients.
# load single cell RNA sequencing data
seurat_obj <- RMedicine2023tidytranscriptomics::seurat_obj
# take a look
seurat_obj## An object of class Seurat
## 3006 features across 33215 samples within 2 assays
## Active assay: SCT (6 features, 0 variable features)
## 1 other assay present: RNA
## 1 dimensional reduction calculated: umaptidyseurat provides a bridge between the Seurat single-cell package and the tidyverse (Wickham et al. 2019). It creates an invisible layer that enables viewing the Seurat object as a tidyverse tibble, and provides Seurat-compatible dplyr, tidyr, ggplot and plotly functions.
If we load the tidyseurat package and then view the single cell data, it now displays as a tibble.
library(tidyseurat)
seurat_obj ## # A Seurat-tibble abstraction: 33,215 × 18
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell file sample_id sample Barcode batch BCB S.Score G2M.Score Phase
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 1_AAACGAA… ../d… SI-GA-H1… SI-GA… AAACGA… 1 BCB1… 0.0620 -0.0972 S
## 2 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0943 -0.193 S
## 3 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0216 -0.124 S
## 4 1_AAAGAAC… ../d… SI-GA-H1… SI-GA… AAAGAA… 1 BCB1… 0.0598 -0.196 S
## 5 1_AAAGGAT… ../d… SI-GA-H1… SI-GA… AAAGGA… 1 BCB1… 0.0281 -0.0540 S
## 6 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0110 -0.0998 S
## 7 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0341 -0.143 S
## 8 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… 0.0425 -0.183 S
## 9 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… -0.0289 -0.127 G1
## 10 1_AAAGTGA… ../d… SI-GA-H1… SI-GA… AAAGTG… 1 BCB1… -0.0551 -0.102 G1
## # ℹ 33,205 more rows
## # ℹ 8 more variables: curated_cell_type <chr>, nCount_RNA <dbl>,
## # nFeature_RNA <int>, nCount_SCT <dbl>, nFeature_SCT <int>, treatment <fct>,
## # UMAP_1 <dbl>, UMAP_2 <dbl>If we want to revert to the standard SingleCellExperiment view we can do that.
options("restore_Seurat_show" = TRUE)
seurat_obj## An object of class Seurat
## 3006 features across 33215 samples within 2 assays
## Active assay: SCT (6 features, 0 variable features)
## 1 other assay present: RNA
## 1 dimensional reduction calculated: umapIf we want to revert back to tidy SingleCellExperiment view we can.
options("restore_Seurat_show" = FALSE)
seurat_obj## # A Seurat-tibble abstraction: 33,215 × 18
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell file sample_id sample Barcode batch BCB S.Score G2M.Score Phase
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 1_AAACGAA… ../d… SI-GA-H1… SI-GA… AAACGA… 1 BCB1… 0.0620 -0.0972 S
## 2 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0943 -0.193 S
## 3 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0216 -0.124 S
## 4 1_AAAGAAC… ../d… SI-GA-H1… SI-GA… AAAGAA… 1 BCB1… 0.0598 -0.196 S
## 5 1_AAAGGAT… ../d… SI-GA-H1… SI-GA… AAAGGA… 1 BCB1… 0.0281 -0.0540 S
## 6 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0110 -0.0998 S
## 7 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0341 -0.143 S
## 8 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… 0.0425 -0.183 S
## 9 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… -0.0289 -0.127 G1
## 10 1_AAAGTGA… ../d… SI-GA-H1… SI-GA… AAAGTG… 1 BCB1… -0.0551 -0.102 G1
## # ℹ 33,205 more rows
## # ℹ 8 more variables: curated_cell_type <chr>, nCount_RNA <dbl>,
## # nFeature_RNA <int>, nCount_SCT <dbl>, nFeature_SCT <int>, treatment <fct>,
## # UMAP_1 <dbl>, UMAP_2 <dbl>It can be interacted with using Seurat
commands such as Assays.
Assays(seurat_obj)## [1] "RNA" "SCT"We can also interact with our object as we do with any tidyverse tibble.
Tidyverse commands
We can use tidyverse commands, such as filter,
select and mutate to explore the tidyseurat
object. Some examples are shown below and more can be seen at the
tidyseurat website here.
We can use filter to choose rows, for example, to see
just the rows for the cells in G1 cell-cycle stage. Check if have groups
or ident present.
seurat_obj |> filter(Phase == "G1")## # A Seurat-tibble abstraction: 18,141 × 18
## # Features=6 | Cells=18141 | Active assay=SCT | Assays=RNA, SCT
## .cell file sample_id sample Barcode batch BCB S.Score G2M.Score Phase
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 1_AAAGTC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… -0.0289 -0.127 G1
## 2 1_AAAGTG… ../d… SI-GA-H1… SI-GA… AAAGTG… 1 BCB1… -0.0551 -0.102 G1
## 3 1_AAATGG… ../d… SI-GA-H1… SI-GA… AAATGG… 1 BCB1… -0.0207 -0.0602 G1
## 4 1_AAATGG… ../d… SI-GA-H1… SI-GA… AAATGG… 1 BCB1… -0.0195 -0.00881 G1
## 5 1_AACAAC… ../d… SI-GA-H1… SI-GA… AACAAC… 1 BCB1… -0.0421 -0.197 G1
## 6 1_AACAAC… ../d… SI-GA-H1… SI-GA… AACAAC… 1 BCB1… -0.0105 -0.0902 G1
## 7 1_AACACA… ../d… SI-GA-H1… SI-GA… AACACA… 1 BCB1… -0.0164 -0.132 G1
## 8 1_AACAGG… ../d… SI-GA-H1… SI-GA… AACAGG… 1 BCB1… -0.00158 -0.0652 G1
## 9 1_AACCAA… ../d… SI-GA-H1… SI-GA… AACCAA… 1 BCB1… -0.00504 -0.135 G1
## 10 1_AACGGG… ../d… SI-GA-H1… SI-GA… AACGGG… 1 BCB1… -0.00701 -0.0919 G1
## # ℹ 18,131 more rows
## # ℹ 8 more variables: curated_cell_type <chr>, nCount_RNA <dbl>,
## # nFeature_RNA <int>, nCount_SCT <dbl>, nFeature_SCT <int>, treatment <fct>,
## # UMAP_1 <dbl>, UMAP_2 <dbl>We can use select to choose columns, for example, to see
the sample, cell, total cellular RNA
seurat_obj |> select(.cell, nCount_RNA, Phase)## # A Seurat-tibble abstraction: 33,215 × 5
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell nCount_RNA Phase UMAP_1 UMAP_2
## <chr> <dbl> <chr> <dbl> <dbl>
## 1 1_AAACGAACAAAGGCAC-1 1485 S 2.56 -2.99
## 2 1_AAACGCTCATGAATAG-1 2460 S -3.32 -3.84
## 3 1_AAACGCTTCTTGGATG-1 1739 S 2.51 -3.15
## 4 1_AAAGAACCAACGGCTC-1 2346 S -3.11 -0.166
## 5 1_AAAGGATTCCCGTTCA-1 763 S -3.50 -0.754
## 6 1_AAAGGGCAGGCATTTC-1 2001 S 1.54 -2.10
## 7 1_AAAGGGCCAGAGATTA-1 1907 S -3.20 -1.92
## 8 1_AAAGTCCAGTAACGAT-1 2239 S -4.03 0.347
## 9 1_AAAGTCCGTAATTAGG-1 1697 G1 -3.24 -3.48
## 10 1_AAAGTGAGTATGGGAC-1 2374 G1 -2.03 -3.56
## # ℹ 33,205 more rowsWe also see the UMAP columns as they are not part of the cell metadata, they are read-only. If we want to save the edited metadata, the Seurat object is modified accordingly.
# Save edited metadata
seurat_modified <- seurat_obj |> select(.cell, nCount_RNA, Phase)
# View Seurat metadata
seurat_modified[[]] |> head()## nCount_RNA Phase
## 1_AAACGAACAAAGGCAC-1 1485 S
## 1_AAACGCTCATGAATAG-1 2460 S
## 1_AAACGCTTCTTGGATG-1 1739 S
## 1_AAAGAACCAACGGCTC-1 2346 S
## 1_AAAGGATTCCCGTTCA-1 763 S
## 1_AAAGGGCAGGCATTTC-1 2001 SWe can use mutate to create a column. For example, we
could create a new Phase_l column that contains a
lower-case version of Phase.
seurat_obj |>
mutate(Phase_l=tolower(Phase)) |>
# Select columns to view
select(.cell, Phase, Phase_l)## # A Seurat-tibble abstraction: 33,215 × 5
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell Phase Phase_l UMAP_1 UMAP_2
## <chr> <chr> <chr> <dbl> <dbl>
## 1 1_AAACGAACAAAGGCAC-1 S s 2.56 -2.99
## 2 1_AAACGCTCATGAATAG-1 S s -3.32 -3.84
## 3 1_AAACGCTTCTTGGATG-1 S s 2.51 -3.15
## 4 1_AAAGAACCAACGGCTC-1 S s -3.11 -0.166
## 5 1_AAAGGATTCCCGTTCA-1 S s -3.50 -0.754
## 6 1_AAAGGGCAGGCATTTC-1 S s 1.54 -2.10
## 7 1_AAAGGGCCAGAGATTA-1 S s -3.20 -1.92
## 8 1_AAAGTCCAGTAACGAT-1 S s -4.03 0.347
## 9 1_AAAGTCCGTAATTAGG-1 G1 g1 -3.24 -3.48
## 10 1_AAAGTGAGTATGGGAC-1 G1 g1 -2.03 -3.56
## # ℹ 33,205 more rowsWe can use tidyverse commands to polish an annotation column. We will extract the sample, and group information from the file name column into separate columns.
# First take a look at the file column
seurat_obj |> select(.cell, file)## # A Seurat-tibble abstraction: 33,215 × 4
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell file UMAP_1 UMAP_2
## <chr> <chr> <dbl> <dbl>
## 1 1_AAACGAACAAAGGCAC-1 ../data/experiment_10X_260819/SI-GA-H1/ou… 2.56 -2.99
## 2 1_AAACGCTCATGAATAG-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -3.32 -3.84
## 3 1_AAACGCTTCTTGGATG-1 ../data/experiment_10X_260819/SI-GA-H1/ou… 2.51 -3.15
## 4 1_AAAGAACCAACGGCTC-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -3.11 -0.166
## 5 1_AAAGGATTCCCGTTCA-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -3.50 -0.754
## 6 1_AAAGGGCAGGCATTTC-1 ../data/experiment_10X_260819/SI-GA-H1/ou… 1.54 -2.10
## 7 1_AAAGGGCCAGAGATTA-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -3.20 -1.92
## 8 1_AAAGTCCAGTAACGAT-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -4.03 0.347
## 9 1_AAAGTCCGTAATTAGG-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -3.24 -3.48
## 10 1_AAAGTGAGTATGGGAC-1 ../data/experiment_10X_260819/SI-GA-H1/ou… -2.03 -3.56
## # ℹ 33,205 more rows
# Create columns for sample and group
seurat_obj <- seurat_obj |>
# Extract sample and group
extract(file, "sample", "../data/.*/([a-zA-Z0-9_-]+)/outs.+", remove = FALSE)
# Take a look
seurat_obj |> select(.cell, sample)## # A Seurat-tibble abstraction: 33,215 × 4
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell sample UMAP_1 UMAP_2
## <chr> <chr> <dbl> <dbl>
## 1 1_AAACGAACAAAGGCAC-1 SI-GA-H1 2.56 -2.99
## 2 1_AAACGCTCATGAATAG-1 SI-GA-H1 -3.32 -3.84
## 3 1_AAACGCTTCTTGGATG-1 SI-GA-H1 2.51 -3.15
## 4 1_AAAGAACCAACGGCTC-1 SI-GA-H1 -3.11 -0.166
## 5 1_AAAGGATTCCCGTTCA-1 SI-GA-H1 -3.50 -0.754
## 6 1_AAAGGGCAGGCATTTC-1 SI-GA-H1 1.54 -2.10
## 7 1_AAAGGGCCAGAGATTA-1 SI-GA-H1 -3.20 -1.92
## 8 1_AAAGTCCAGTAACGAT-1 SI-GA-H1 -4.03 0.347
## 9 1_AAAGTCCGTAATTAGG-1 SI-GA-H1 -3.24 -3.48
## 10 1_AAAGTGAGTATGGGAC-1 SI-GA-H1 -2.03 -3.56
## # ℹ 33,205 more rowsWe could use tidyverse unite to combine columns, for
example to create a new column for sample id that combines the sample
and patient identifier (BCB) columns.
seurat_obj <- seurat_obj |> unite("sample_id", sample, BCB, remove = FALSE)
# Take a look
seurat_obj |> select(.cell, sample_id, sample, BCB)## # A Seurat-tibble abstraction: 33,215 × 6
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell sample_id sample BCB UMAP_1 UMAP_2
## <chr> <chr> <chr> <chr> <dbl> <dbl>
## 1 1_AAACGAACAAAGGCAC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 2.56 -2.99
## 2 1_AAACGCTCATGAATAG-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.32 -3.84
## 3 1_AAACGCTTCTTGGATG-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 2.51 -3.15
## 4 1_AAAGAACCAACGGCTC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.11 -0.166
## 5 1_AAAGGATTCCCGTTCA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.50 -0.754
## 6 1_AAAGGGCAGGCATTTC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 1.54 -2.10
## 7 1_AAAGGGCCAGAGATTA-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.20 -1.92
## 8 1_AAAGTCCAGTAACGAT-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -4.03 0.347
## 9 1_AAAGTCCGTAATTAGG-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -3.24 -3.48
## 10 1_AAAGTGAGTATGGGAC-1 SI-GA-H1_BCB102 SI-GA-H1 BCB102 -2.03 -3.56
## # ℹ 33,205 more rowsPart 2 Signature visualisation
We will now demonstrate a real-world example of the power of using tidy transcriptomics packages in single cell analysis. For more information on single-cell analysis steps performed in a tidy way please see the ISMB2021 workshop.
Data pre-processing
The object seurat_obj we’ve been using was created as
part of a study on breast cancer systemic immune response. Peripheral
blood mononuclear cells have been sequenced for RNA at the single-cell
level. The steps used to generate the object are summarised below.
scran,scater, andDropletsUtilspackages have been used to eliminate empty droplets and dead cells. Samples were individually quality checked and cells were filtered for good gene coverage.Variable features were identified using
Seurat.Read counts were scaled and normalised using SCTtransform from
Seurat.Data integration was performed using
Seuratwith default parameters.PCA performed to reduce feature dimensionality.
Nearest-neighbor cell networks were calculated using 30 principal components.
2 UMAP dimensions were calculated using 30 principal components.
Cells with similar transcriptome profiles were grouped into clusters using Louvain clustering from
Seurat.
Analyse custom signature
The researcher analysing this dataset wanted to to identify gamma delta T cells using a gene signature from a published paper (Pizzolato et al. 2019).
With tidyseurat’s join_features the counts for the genes
could be viewed as columns.
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) ## # A Seurat-tibble abstraction: 33,215 × 24
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell file sample_id sample Barcode batch BCB S.Score G2M.Score Phase
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 1_AAACGAA… ../d… SI-GA-H1… SI-GA… AAACGA… 1 BCB1… 0.0620 -0.0972 S
## 2 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0943 -0.193 S
## 3 1_AAACGCT… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0216 -0.124 S
## 4 1_AAAGAAC… ../d… SI-GA-H1… SI-GA… AAAGAA… 1 BCB1… 0.0598 -0.196 S
## 5 1_AAAGGAT… ../d… SI-GA-H1… SI-GA… AAAGGA… 1 BCB1… 0.0281 -0.0540 S
## 6 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0110 -0.0998 S
## 7 1_AAAGGGC… ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0341 -0.143 S
## 8 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… 0.0425 -0.183 S
## 9 1_AAAGTCC… ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… -0.0289 -0.127 G1
## 10 1_AAAGTGA… ../d… SI-GA-H1… SI-GA… AAAGTG… 1 BCB1… -0.0551 -0.102 G1
## # ℹ 33,205 more rows
## # ℹ 14 more variables: curated_cell_type <chr>, nCount_RNA <dbl>,
## # nFeature_RNA <int>, nCount_SCT <dbl>, nFeature_SCT <int>, treatment <fct>,
## # CD3D <dbl>, TRDC <dbl>, TRGC1 <dbl>, TRGC2 <dbl>, CD8A <dbl>, CD8B <dbl>,
## # UMAP_1 <dbl>, UMAP_2 <dbl>Signature calculation
They were able to use tidyseurat’s join_features to
select the counts for the genes in the signature, followed by tidyverse
mutate to easily create a column containing the signature
score.
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) |>
mutate(signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to=c(0,1)) -
scales::rescale(CD8A + CD8B, to=c(0,1))
) |>
select(signature_score, everything())## # A Seurat-tibble abstraction: 33,215 × 25
## # Features=6 | Cells=33215 | Active assay=SCT | Assays=RNA, SCT
## .cell signature_score file sample_id sample Barcode batch BCB S.Score
## <chr> <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <dbl>
## 1 1_AAACGAA… 0.165 ../d… SI-GA-H1… SI-GA… AAACGA… 1 BCB1… 0.0620
## 2 1_AAACGCT… 0.0647 ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0943
## 3 1_AAACGCT… 0.241 ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0216
## 4 1_AAAGAAC… 0.104 ../d… SI-GA-H1… SI-GA… AAAGAA… 1 BCB1… 0.0598
## 5 1_AAAGGAT… 0.165 ../d… SI-GA-H1… SI-GA… AAAGGA… 1 BCB1… 0.0281
## 6 1_AAAGGGC… 0.208 ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0110
## 7 1_AAAGGGC… 0 ../d… SI-GA-H1… SI-GA… AAAGGG… 1 BCB1… 0.0341
## 8 1_AAAGTCC… 0.104 ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… 0.0425
## 9 1_AAAGTCC… 0.165 ../d… SI-GA-H1… SI-GA… AAAGTC… 1 BCB1… -0.0289
## 10 1_AAAGTGA… 0.165 ../d… SI-GA-H1… SI-GA… AAAGTG… 1 BCB1… -0.0551
## # ℹ 33,205 more rows
## # ℹ 16 more variables: G2M.Score <dbl>, Phase <chr>, curated_cell_type <chr>,
## # nCount_RNA <dbl>, nFeature_RNA <int>, nCount_SCT <dbl>, nFeature_SCT <int>,
## # treatment <fct>, CD3D <dbl>, TRDC <dbl>, TRGC1 <dbl>, TRGC2 <dbl>,
## # CD8A <dbl>, CD8B <dbl>, UMAP_1 <dbl>, UMAP_2 <dbl>The gamma delta T cells could then be visualised by the signature score using Seurat’s visualisation functions.
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) |>
mutate(signature_score =
scales::rescale(CD3D + TRDC + TRGC1+ TRGC2, to=c(0,1)) -
scales::rescale(CD8A + CD8B, to=c(0,1))
) |>
Seurat::FeaturePlot(features = "signature_score", min.cutoff = 0) 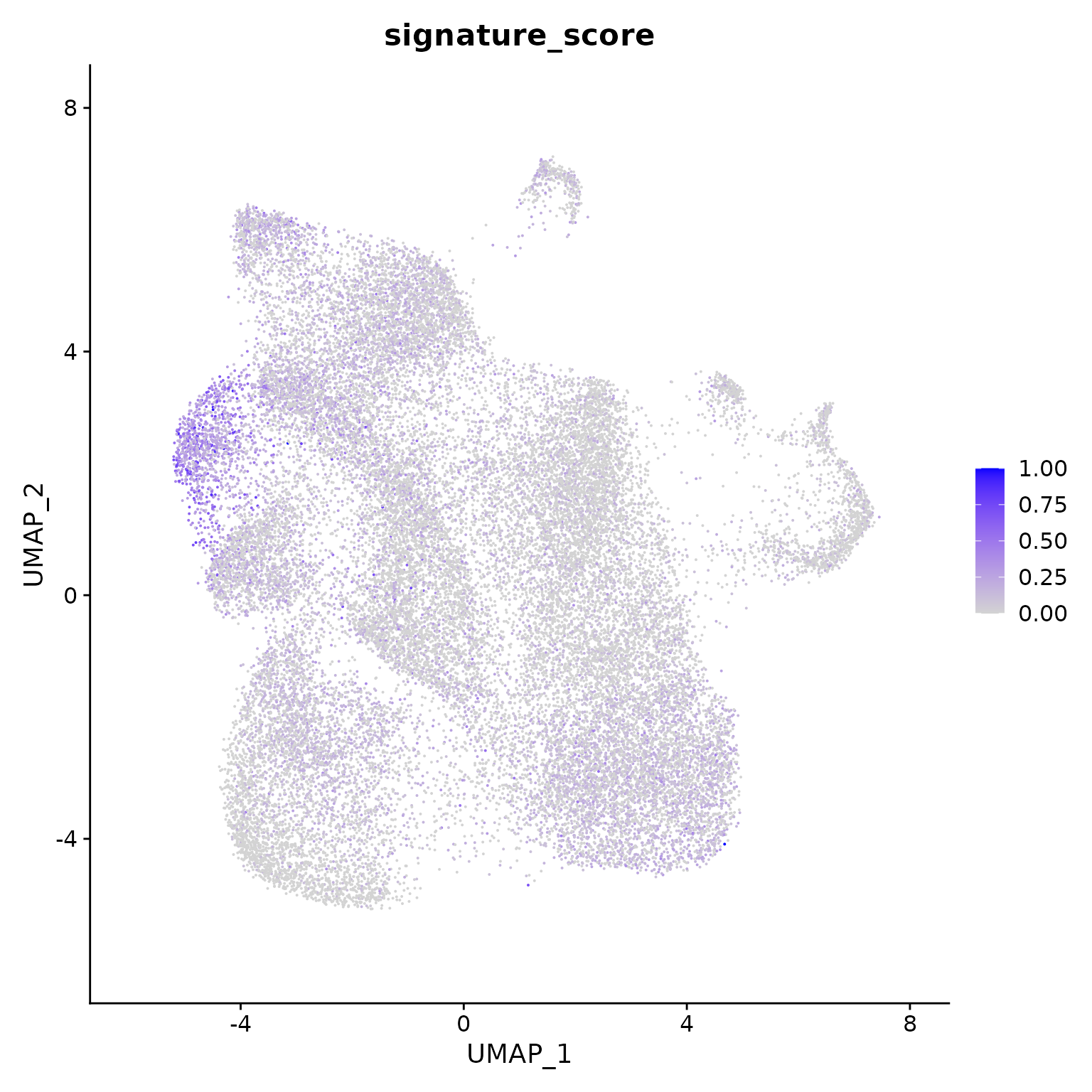
The cells could also be visualised using the popular and powerful ggplot package, enabling the researcher to use ggplot functions they were familiar with, and to customise the plot with great flexibility.
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) |>
mutate(signature_score =
scales::rescale(CD3D + TRDC + TRGC1+ TRGC2, to=c(0,1)) -
scales::rescale(CD8A + CD8B, to=c(0,1))
) |>
# plot cells with high score last so they're not obscured by other cells
arrange(signature_score) |>
ggplot(aes(UMAP_1, UMAP_2, color = signature_score)) +
geom_point(size=0.5) +
scale_color_distiller(palette = "Spectral") +
RMedicine2023tidytranscriptomics::theme_multipanel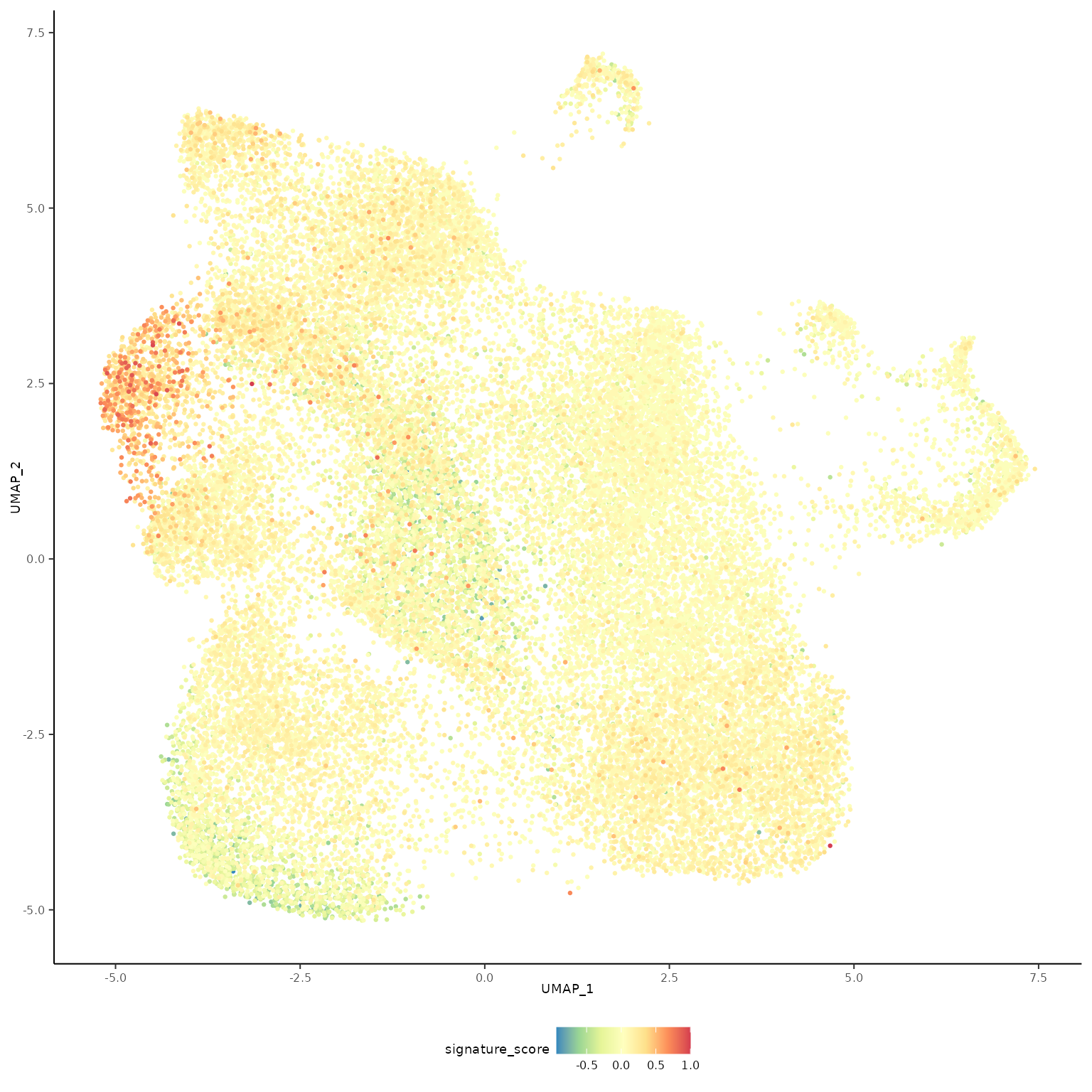
Filtering based on gate
The gamma delta T cells (the blue cluster on the left with high signature score) could be interactively selected from the plot using the tidygate package.
This code can only be executed interactively from an R file and not from a markdown file. So, in this casre, we need to copy and paste this into the console.
seurat_obj_gamma_delta =
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) |>
mutate(signature_score =
scales::rescale(CD3D + TRDC + TRGC1+ TRGC2, to=c(0,1)) -
scales::rescale(CD8A + CD8B, to=c(0,1))
) |>
mutate(gate = tidygate::gate_int(
UMAP_1, UMAP_2,
.size = 0.1,
.color =signature_score
)) |>
filter(gate==1)Filtering based on threshold
For exploratory analyses, we can select the gamma delta T cells, the red cluster on the left with high signature score. We’ll filter for cells with a signature score > 0.7.
seurat_obj_gamma_delta <-
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"), shape = "wide"
) |>
mutate(
signature_score =
scales::rescale(CD3D + TRDC + TRGC1 + TRGC2, to = c(0, 1)) -
scales::rescale(CD8A + CD8B, to = c(0, 1))
) |>
# Proper cluster selection should be used instead (see supplementary material)
filter(signature_score > 0.5)Reanalysis after filtering
It was then possible to perform analyses on these gamma delta T cells by simply chaining further commands, such as below.
source("https://raw.githubusercontent.com/satijalab/seurat-wrappers/master/R/fast_mnn.R")
seurat_obj |>
join_features(
features = c("CD3D", "TRDC", "TRGC1", "TRGC2", "CD8A", "CD8B"),
shape = "wide",
assay = "SCT"
) |>
mutate(signature_score =
scales::rescale(CD3D + TRDC + TRGC1+ TRGC2, to=c(0,1)) -
scales::rescale(CD8A + CD8B, to=c(0,1))
) |>
# Proper cluster selection should be used instead (see supplementary material)
filter(signature_score > 0.5) |>
# Reanalysis
NormalizeData(assay="RNA") |>
FindVariableFeatures(nfeatures = 100, assay="RNA") |>
SplitObject(split.by = "file") |>
SeuratWrappers::RunFastMNN(assay="RNA", features = 100) |>
RunUMAP(reduction = "mnn", dims = 1:20) |>
FindNeighbors(dims = 1:20, reduction = "mnn") |>
FindClusters(resolution = 0.3)For comparison, we show the alternative using base R and SingleCellExperiment. Note that the code contains more redundancy and intermediate objects.
counts_positive <-
assay(seurat_obj, "logcounts")[c("CD3D", "TRDC", "TRGC1", "TRGC2"), ] |>
colSums() |>
scales::rescale(to = c(0, 1))
counts_negative <-
assay(seurat_obj, "logcounts")[c("CD8A", "CD8B"), ] |>
colSums() |>
scales::rescale(to = c(0, 1))
seurat_obj$signature_score <- counts_positive - counts_negative
seurat_obj_gamma_delta <- seurat_obj[, seurat_obj$signature_score > 0.7]
# Reanalysis
# ...Interactive plotting
As a final note, it’s also possible to do complex and powerful things in a simple way, due to the integration of the tidy transcriptomics packages with the tidy universe. As one example, we can visualise the cells as a 3D plot using plotly.
The example data we’ve been using only contains a few genes, for the sake of time and size in this demonstration, but below is how you could generate the 3 dimensions needed for 3D plot with a full dataset.
single_cell_object |>
RunUMAP(dims = 1:30, n.components = 3L, spread = 0.5,min.dist = 0.01, n.neighbors = 10L)We’ll demonstrate creating a 3D plot using some data that has 3 UMAP dimensions.
seurat_obj_UMAP3 <- RMedicine2023tidytranscriptomics::seurat_obj_UMAP3
# Look at the object
seurat_obj_UMAP3 |> select(contains("UMAP"), everything())## # A Seurat-tibble abstraction: 33,215 × 17
## # Features=1 | Cells=33215 | Active assay=integrated | Assays=integrated
## .cell file Barcode batch BCB S.Score G2M.Score Phase curated_cell_type
## <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr>
## 1 1_AAACGA… ../d… AAACGA… 1 BCB1… 0.0620 -0.0972 S CD4+_Tcm_S100A4_…
## 2 1_AAACGC… ../d… AAACGC… 1 BCB1… 0.0943 -0.193 S CD8+_high_ribonu…
## 3 1_AAACGC… ../d… AAACGC… 1 BCB1… 0.0216 -0.124 S CD4+_Tcm_S100A4_…
## 4 1_AAAGAA… ../d… AAAGAA… 1 BCB1… 0.0598 -0.196 S MAIT
## 5 1_AAAGGA… ../d… AAAGGA… 1 BCB1… 0.0281 -0.0540 S CD8+_high_ribonu…
## 6 1_AAAGGG… ../d… AAAGGG… 1 BCB1… 0.0110 -0.0998 S MAIT
## 7 1_AAAGGG… ../d… AAAGGG… 1 BCB1… 0.0341 -0.143 S CD8+_high_ribonu…
## 8 1_AAAGTC… ../d… AAAGTC… 1 BCB1… 0.0425 -0.183 S MAIT
## 9 1_AAAGTC… ../d… AAAGTC… 1 BCB1… -0.0289 -0.127 G1 CD8+_high_ribonu…
## 10 1_AAAGTG… ../d… AAAGTG… 1 BCB1… -0.0551 -0.102 G1 CD8+_high_ribonu…
## # ℹ 33,205 more rows
## # ℹ 8 more variables: PC_1 <dbl>, PC_2 <dbl>, PC_3 <dbl>, PC_4 <dbl>,
## # PC_5 <dbl>, UMAP_1 <dbl>, UMAP_2 <dbl>, UMAP_3 <dbl>
seurat_obj_UMAP3 |>
tidyseurat::plot_ly(
x = ~`UMAP_1`,
y = ~`UMAP_2`,
z = ~`UMAP_3`,
color = ~curated_cell_type,
colors = dittoSeq::dittoColors()
) |>
plotly::add_markers(size = I(1))Part 3 Nested analyses
When analysing single cell data is sometimes necessary to perform calculations on data subsets. For example, we might want to estimate difference in mRNA abundance between two condition for each cell type.
tidyr and purrr offer a great tool to
perform iterativre analyses in a functional way.
We use tidyverse nest to group the data. The command
below will create a tibble containing a column with a
SummarizedExperiment object for each cell type. nest is
similar to tidyverse group_by, except with
nest each group is stored in a single row, and can be a
complex object such as Seurat.
First let’s have a look to the cell types that constitute this dataset
seurat_obj |>
count(curated_cell_type)## tidyseurat says: A data frame is returned for independent data analysis.## # A tibble: 13 × 2
## curated_cell_type n
## <chr> <int>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM 4978
## 2 CD8+_Tcm 3611
## 3 CD8+_Tem 4157
## 4 CD8+_high_ribonucleosome 4885
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 4138
## 6 HSC_lymphoid_myeloid_like 1299
## 7 MAIT 1909
## 8 NK_cells 4535
## 9 NK_cycling 329
## 10 TCR_V_Delta_1 651
## 11 TCR_V_Delta_2 355
## 12 T_cell:CD8+_other 2109
## 13 limphoid_myeloid_like_HLA_CD74 259Let’s group the cells based on cell identity using
nest
# Set idents for the differential analysis
Idents(seurat_obj) = seurat_obj[[]]$"treatment"
seurat_obj_nested =
seurat_obj |>
nest(seurat = -curated_cell_type)
seurat_obj_nested## # A tibble: 13 × 2
## curated_cell_type seurat
## <chr> <list>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <Seurat[,4978]>
## 2 CD8+_high_ribonucleosome <Seurat[,4885]>
## 3 MAIT <Seurat[,1909]>
## 4 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <Seurat[,4138]>
## 5 TCR_V_Delta_2 <Seurat[,355]>
## 6 T_cell:CD8+_other <Seurat[,2109]>
## 7 NK_cells <Seurat[,4535]>
## 8 CD8+_Tem <Seurat[,4157]>
## 9 CD8+_Tcm <Seurat[,3611]>
## 10 TCR_V_Delta_1 <Seurat[,651]>
## 11 NK_cycling <Seurat[,329]>
## 12 HSC_lymphoid_myeloid_like <Seurat[,1299]>
## 13 limphoid_myeloid_like_HLA_CD74 <Seurat[,259]>Let’s see what the first element of the Surat column looks like
## [[1]]
## # A Seurat-tibble abstraction: 4,978 × 17
## # Features=6 | Cells=4978 | Active assay=SCT | Assays=RNA, SCT
## .cell file sample_id sample Barcode batch BCB S.Score G2M.Score Phase
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 1_AAACGA… ../d… SI-GA-H1… SI-GA… AAACGA… 1 BCB1… 0.0620 -0.0972 S
## 2 1_AAACGC… ../d… SI-GA-H1… SI-GA… AAACGC… 1 BCB1… 0.0216 -0.124 S
## 3 1_AAATGG… ../d… SI-GA-H1… SI-GA… AAATGG… 1 BCB1… -0.0195 -0.00881 G1
## 4 1_AACAAA… ../d… SI-GA-H1… SI-GA… AACAAA… 1 BCB1… 0.0336 -0.147 S
## 5 1_AACAAC… ../d… SI-GA-H1… SI-GA… AACAAC… 1 BCB1… -0.0421 -0.197 G1
## 6 1_AACAAC… ../d… SI-GA-H1… SI-GA… AACAAC… 1 BCB1… -0.0105 -0.0902 G1
## 7 1_AACACA… ../d… SI-GA-H1… SI-GA… AACACA… 1 BCB1… 0.00198 -0.0542 S
## 8 1_AACCAA… ../d… SI-GA-H1… SI-GA… AACCAA… 1 BCB1… -0.00504 -0.135 G1
## 9 1_AACCAA… ../d… SI-GA-H1… SI-GA… AACCAA… 1 BCB1… 0.0704 -0.145 S
## 10 1_AACTTC… ../d… SI-GA-H1… SI-GA… AACTTC… 1 BCB1… -0.0336 -0.0956 G1
## # ℹ 4,968 more rows
## # ℹ 7 more variables: nCount_RNA <dbl>, nFeature_RNA <int>, nCount_SCT <dbl>,
## # nFeature_SCT <int>, treatment <fct>, UMAP_1 <dbl>, UMAP_2 <dbl>Now, let’s perform a differential gene-transcript abundance analysis between the two conditions for each cell type.
seurat_obj_nested =
seurat_obj_nested |>
# Select significant genes
mutate(significant_genes = map(
seurat,
~ .x |>
# Test
NormalizeData(assay="RNA") |>
FindAllMarkers(assay="RNA") |>
# Select top genes
filter(p_val_adj<0.05) |>
head(10) |>
rownames()
))
seurat_obj_nested## # A tibble: 13 × 3
## curated_cell_type seurat significant_genes
## <chr> <list> <list>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <Seurat[,4978]> <chr [10]>
## 2 CD8+_high_ribonucleosome <Seurat[,4885]> <chr [10]>
## 3 MAIT <Seurat[,1909]> <chr [10]>
## 4 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <Seurat[,4138]> <chr [10]>
## 5 TCR_V_Delta_2 <Seurat[,355]> <chr [10]>
## 6 T_cell:CD8+_other <Seurat[,2109]> <chr [10]>
## 7 NK_cells <Seurat[,4535]> <chr [10]>
## 8 CD8+_Tem <Seurat[,4157]> <chr [10]>
## 9 CD8+_Tcm <Seurat[,3611]> <chr [10]>
## 10 TCR_V_Delta_1 <Seurat[,651]> <chr [10]>
## 11 NK_cycling <Seurat[,329]> <chr [10]>
## 12 HSC_lymphoid_myeloid_like <Seurat[,1299]> <chr [10]>
## 13 limphoid_myeloid_like_HLA_CD74 <Seurat[,259]> <chr [10]>We can the lies the top genes with the heat map iteratively across the cell types
seurat_obj_nested =
seurat_obj_nested |>
# Build heatmaps
mutate(heatmap = map2(
seurat, significant_genes,
~ .x |>
ScaleData(assay="RNA") |>
DoHeatmap(.y, assay="RNA")
))
seurat_obj_nested## # A tibble: 13 × 4
## curated_cell_type seurat significant_genes heatmap
## <chr> <list> <list> <list>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <Seurat[,4978]> <chr [10]> <patchwrk>
## 2 CD8+_high_ribonucleosome <Seurat[,4885]> <chr [10]> <patchwrk>
## 3 MAIT <Seurat[,1909]> <chr [10]> <patchwrk>
## 4 CD8+_transitional_effector_GZMK… <Seurat[,4138]> <chr [10]> <patchwrk>
## 5 TCR_V_Delta_2 <Seurat[,355]> <chr [10]> <patchwrk>
## 6 T_cell:CD8+_other <Seurat[,2109]> <chr [10]> <patchwrk>
## 7 NK_cells <Seurat[,4535]> <chr [10]> <patchwrk>
## 8 CD8+_Tem <Seurat[,4157]> <chr [10]> <patchwrk>
## 9 CD8+_Tcm <Seurat[,3611]> <chr [10]> <patchwrk>
## 10 TCR_V_Delta_1 <Seurat[,651]> <chr [10]> <patchwrk>
## 11 NK_cycling <Seurat[,329]> <chr [10]> <patchwrk>
## 12 HSC_lymphoid_myeloid_like <Seurat[,1299]> <chr [10]> <patchwrk>
## 13 limphoid_myeloid_like_HLA_CD74 <Seurat[,259]> <chr [10]> <patchwrk>Let’s have a look to the first heatmap
## [[1]]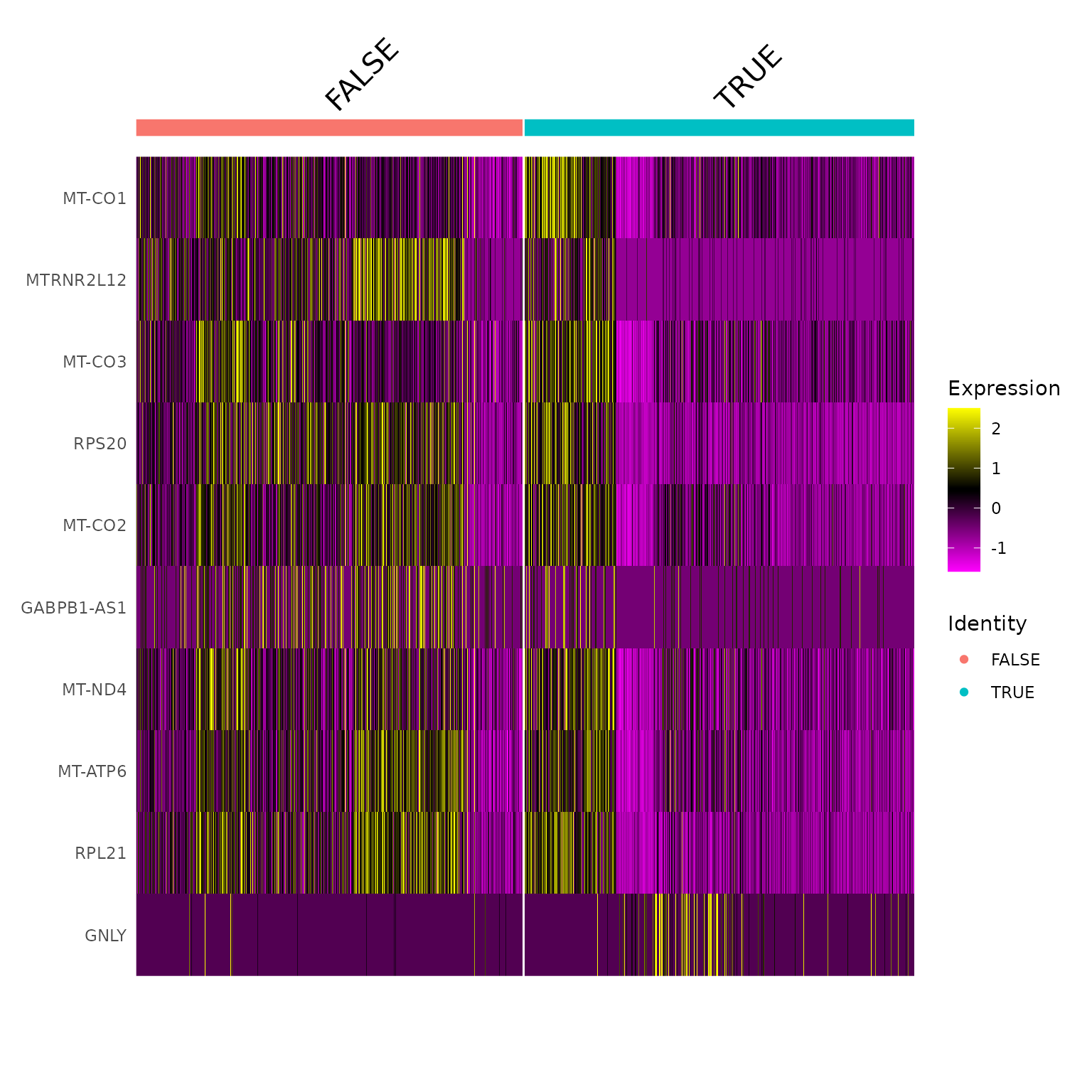
You can do this whole analysis without saving any temporary variable using the piping functionality of tidy R programming
seurat_obj |>
# Nest
nest(seurat = -curated_cell_type) |>
# Select significant genes
mutate(significant_genes = map(
seurat,
~ .x |>
# Test
NormalizeData(assay="RNA") |>
FindAllMarkers(assay="RNA") |>
# Select top genes
filter(p_val_adj<0.05) |>
head(10) |>
rownames()
)) |>
# Build heatmaps
mutate(heatmap = map2(
seurat, significant_genes,
~ .x |>
ScaleData(assay="RNA") |>
DoHeatmap(.y, assay="RNA")
)) |>
# Extract heatmaps
pull(heatmap)Exercises
- Let’s suppose that you want to perform the analyses only for cell types that have a total number of cells bigger than 1000. For example, if a cell type has less than a sum of 1000 cells across all samples, that cell type will be dropped from the dataset.
- Answer this question avoiding to save temporary variables, and using the function add_count to count the cells (before nesting), and then filter
- Answer this question avoiding to save temporary variables, and using the function map_int to count the cells (after nesting), and the filter
Part 4 Pseudobulk analyses
Next we want to identify genes whose transcription is affected by treatment in this dataset, comparing treated and untreated patients. We can do this with pseudobulk analysis. We aggregate cell-wise transcript abundance into pseudobulk samples and can then perform hypothesis testing using the very well established bulk RNA sequencing tools. For example, we can use DESeq2 in tidybulk to perform differential expression testing. For more details on pseudobulk analysis see here.
We want to do it for each cell type and the tidy transcriptomics ecosystem makes this very easy.
Create pseudobulk samples
To create pseudobulk samples from the single cell samples, we will
use a helper function called aggregate_cells, available in
this workshop package. This function will combine the single cells into
a group for each cell type for each sample.
pseudo_bulk <-
seurat_obj |>
RMedicine2023tidytranscriptomics::aggregate_cells(c(sample, curated_cell_type), assays = "RNA")
pseudo_bulk## # A SummarizedExperiment-tibble abstraction: 369,000 × 12
## # Features=3000 | Samples=123 | Assays=RNA
## .feature .sample RNA sample curated_cell_type .aggregated_cells file
## <chr> <chr> <dbl> <chr> <chr> <int> <chr>
## 1 A1BG-AS1 SI-GA-E5___C… 1 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 2 A2M-AS1 SI-GA-E5___C… 14 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 3 AAED1 SI-GA-E5___C… 18 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 4 ABCA2 SI-GA-E5___C… 4 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 5 ABCB1 SI-GA-E5___C… 11 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 6 ABCC4 SI-GA-E5___C… 4 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 7 ABHD15 SI-GA-E5___C… 6 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 8 ABHD17A SI-GA-E5___C… 50 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 9 ABHD17B SI-GA-E5___C… 6 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## 10 ABHD2 SI-GA-E5___C… 12 SI-GA… CD4+_Tcm_S100A4_… 372 ../d…
## # ℹ 40 more rows
## # ℹ 5 more variables: sample_id <chr>, batch <chr>, BCB <chr>, treatment <fct>,
## # feature <chr>Tidybulk and tidySummarizedExperiment
With tidySummarizedExperiment and tidybulk
it is easy to split the data into groups and perform analyses on each
without needing to create separate objects.
Tidybulk functions/utilities available
| Function | Description |
|---|---|
aggregate_duplicates |
Aggregate abundance and annotation of duplicated transcripts in a robust way |
identify_abundant keep_abundant
|
identify or keep the abundant genes |
keep_variable |
Filter for top variable features |
scale_abundance |
Scale (normalise) abundance for RNA sequencing depth |
reduce_dimensions |
Perform dimensionality reduction (PCA, MDS, tSNE, UMAP) |
cluster_elements |
Labels elements with cluster identity (kmeans, SNN) |
remove_redundancy |
Filter out elements with highly correlated features |
adjust_abundance |
Remove known unwanted variation (Combat) |
test_differential_abundance |
Differential transcript abundance testing (DESeq2, edgeR, voom) |
deconvolve_cellularity |
Estimated tissue composition (Cibersort, llsr, epic, xCell, mcp_counter, quantiseq |
test_differential_cellularity |
Differential cell-type abundance testing |
test_stratification_cellularity |
Estimate Kaplan-Meier survival differences |
test_gene_enrichment |
Gene enrichment analyses (EGSEA) |
test_gene_overrepresentation |
Gene enrichment on list of transcript names (no rank) |
test_gene_rank |
Gene enrichment on list of transcript (GSEA) |
impute_missing_abundance |
Impute abundance for missing data points using sample groupings |
We use tidyverse nest to group the data. The command
below will create a tibble containing a column with a
SummarizedExperiment object for each cell type. nest is
similar to tidyverse group_by, except with
nest each group is stored in a single row, and can be a
complex object such as a plot or SummarizedExperiment.
pseudo_bulk_nested <-
pseudo_bulk |>
nest(grouped_summarized_experiment = -curated_cell_type)
pseudo_bulk_nested## # A tibble: 13 × 2
## curated_cell_type grouped_summarized_experiment
## <chr> <list>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]>
## 2 CD8+_Tcm <SmmrzdEx[,10]>
## 3 CD8+_Tem <SmmrzdEx[,10]>
## 4 CD8+_high_ribonucleosome <SmmrzdEx[,10]>
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <SmmrzdEx[,10]>
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]>
## 7 MAIT <SmmrzdEx[,10]>
## 8 NK_cells <SmmrzdEx[,10]>
## 9 NK_cycling <SmmrzdEx[,10]>
## 10 TCR_V_Delta_1 <SmmrzdEx[,10]>
## 11 TCR_V_Delta_2 <SmmrzdEx[,9]>
## 12 T_cell:CD8+_other <SmmrzdEx[,10]>
## 13 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,4]>To explore the grouping, we can use tidyverse slice to
choose a row (cell_type) and pull to extract the values
from a column. If we pull the data column we can view the
SummarizedExperiment object.
## [[1]]
## # A SummarizedExperiment-tibble abstraction: 30,000 × 11
## # Features=3000 | Samples=10 | Assays=RNA
## .feature .sample RNA sample .aggregated_cells file sample_id batch BCB
## <chr> <chr> <dbl> <chr> <int> <chr> <chr> <chr> <chr>
## 1 A1BG-AS1 SI-GA-E5… 1 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 2 A2M-AS1 SI-GA-E5… 14 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 3 AAED1 SI-GA-E5… 18 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 4 ABCA2 SI-GA-E5… 4 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 5 ABCB1 SI-GA-E5… 11 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 6 ABCC4 SI-GA-E5… 4 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 7 ABHD15 SI-GA-E5… 6 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 8 ABHD17A SI-GA-E5… 50 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 9 ABHD17B SI-GA-E5… 6 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## 10 ABHD2 SI-GA-E5… 12 SI-GA… 372 ../d… SI-GA-E5… 2 BCB0…
## # ℹ 40 more rows
## # ℹ 2 more variables: treatment <fct>, feature <chr>We can then identify differentially expressed genes for each cell
type for our condition of interest, treated versus untreated patients.
We use tidyverse map to apply differential expression
functions to each cell type group in the nested data. The result columns
will be added to the SummarizedExperiment objects.
# Differential transcription abundance
pseudo_bulk_nested <-
pseudo_bulk_nested |>
# map accepts a data column (.x) and a function. It applies the function to each element of the column.
mutate(grouped_summarized_experiment = map(
grouped_summarized_experiment,
~ .x |>
# Removing genes with low expression
keep_abundant(factor_of_interest = treatment) |>
# Testing for differential expression using DESeq2
test_differential_abundance(~treatment, method="DESeq2") |>
# Scale abundance for FUTURE visualisation
scale_abundance(method="TMMwsp")
))The output is again a tibble containing a SummarizedExperiment object for each cell type.
pseudo_bulk_nested## # A tibble: 13 × 2
## curated_cell_type grouped_summarized_experiment
## <chr> <list>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]>
## 2 CD8+_Tcm <SmmrzdEx[,10]>
## 3 CD8+_Tem <SmmrzdEx[,10]>
## 4 CD8+_high_ribonucleosome <SmmrzdEx[,10]>
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <SmmrzdEx[,10]>
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]>
## 7 MAIT <SmmrzdEx[,10]>
## 8 NK_cells <SmmrzdEx[,10]>
## 9 NK_cycling <SmmrzdEx[,10]>
## 10 TCR_V_Delta_1 <SmmrzdEx[,10]>
## 11 TCR_V_Delta_2 <SmmrzdEx[,9]>
## 12 T_cell:CD8+_other <SmmrzdEx[,10]>
## 13 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,4]>If we pull out the SummarizedExperiment object for the first cell type, as before, we can see it now has columns containing the differential expression results (e.g. logFC, PValue).
## [[1]]
## # A SummarizedExperiment-tibble abstraction: 14,580 × 21
## # Features=1458 | Samples=10 | Assays=RNA, RNA_scaled
## .feature .sample RNA RNA_scaled sample .aggregated_cells file sample_id
## <chr> <chr> <dbl> <dbl> <chr> <int> <chr> <chr>
## 1 A2M-AS1 SI-GA-E5_… 14 56.3 SI-GA… 372 ../d… SI-GA-E5…
## 2 AAED1 SI-GA-E5_… 18 72.4 SI-GA… 372 ../d… SI-GA-E5…
## 3 ABCA2 SI-GA-E5_… 4 16.1 SI-GA… 372 ../d… SI-GA-E5…
## 4 ABCB1 SI-GA-E5_… 11 44.2 SI-GA… 372 ../d… SI-GA-E5…
## 5 ABHD15 SI-GA-E5_… 6 24.1 SI-GA… 372 ../d… SI-GA-E5…
## 6 ABHD17A SI-GA-E5_… 50 201. SI-GA… 372 ../d… SI-GA-E5…
## 7 ABHD17B SI-GA-E5_… 6 24.1 SI-GA… 372 ../d… SI-GA-E5…
## 8 ABHD2 SI-GA-E5_… 12 48.3 SI-GA… 372 ../d… SI-GA-E5…
## 9 ABHD5 SI-GA-E5_… 5 20.1 SI-GA… 372 ../d… SI-GA-E5…
## 10 ABI2 SI-GA-E5_… 22 88.5 SI-GA… 372 ../d… SI-GA-E5…
## # ℹ 40 more rows
## # ℹ 13 more variables: batch <chr>, BCB <chr>, treatment <fct>, TMM <dbl>,
## # multiplier <dbl>, feature <chr>, .abundant <lgl>, baseMean <dbl>,
## # log2FoldChange <dbl>, lfcSE <dbl>, stat <dbl>, pvalue <dbl>, padj <dbl>We can analyse our nested dataset mapping queries across the
SummarizedExperiments
pseudo_bulk_nested =
pseudo_bulk_nested |>
# Identify top significant genes
mutate(top_genes = map_chr(
grouped_summarized_experiment,
~ .x |>
pivot_transcript() |>
arrange(pvalue) |>
head(1) |>
pull(.feature)
)) |>
# Filter top gene
mutate(grouped_summarized_experiment = map2(
grouped_summarized_experiment, top_genes,
~ filter(.x, .feature == .y)
))
pseudo_bulk_nested## # A tibble: 13 × 3
## curated_cell_type grouped_summarized_ex…¹ top_genes
## <chr> <list> <chr>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]> MIAT
## 2 CD8+_Tcm <SmmrzdEx[,10]> SH3BGRL3
## 3 CD8+_Tem <SmmrzdEx[,10]> MT-CO1
## 4 CD8+_high_ribonucleosome <SmmrzdEx[,10]> PPBP
## 5 CD8+_transitional_effector_GZMK_KLRB1_LYAR4 <SmmrzdEx[,10]> TYROBP
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]> GABPB1-A…
## 7 MAIT <SmmrzdEx[,10]> MT-CO1
## 8 NK_cells <SmmrzdEx[,10]> MT-CO1
## 9 NK_cycling <SmmrzdEx[,10]> CD3G
## 10 TCR_V_Delta_1 <SmmrzdEx[,10]> PRF1
## 11 TCR_V_Delta_2 <SmmrzdEx[,9]> MT-CO1
## 12 T_cell:CD8+_other <SmmrzdEx[,10]> CTSW
## 13 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,4]> GNLY
## # ℹ abbreviated name: ¹grouped_summarized_experimentPlot top differential genes
pseudo_bulk_nested =
pseudo_bulk_nested |>
# Plot significant genes for each cell type
# map2 is map that accepts 2 input columns (.x, .y) and a function
mutate(plot = map2(
grouped_summarized_experiment,curated_cell_type,
~ .x |>
# Plot
ggplot(aes(treatment, RNA_scaled + 1)) +
geom_boxplot(aes(fill = treatment)) +
geom_jitter() +
scale_y_log10() +
facet_wrap(~.feature, ncol = 3) +
ggtitle(.y) +
RMedicine2023tidytranscriptomics::theme_multipanel
))
pseudo_bulk_nested## # A tibble: 13 × 4
## curated_cell_type grouped_summarized_e…¹ top_genes plot
## <chr> <list> <chr> <lis>
## 1 CD4+_Tcm_S100A4_IL32_IL7R_VIM <SmmrzdEx[,10]> MIAT <gg>
## 2 CD8+_Tcm <SmmrzdEx[,10]> SH3BGRL3 <gg>
## 3 CD8+_Tem <SmmrzdEx[,10]> MT-CO1 <gg>
## 4 CD8+_high_ribonucleosome <SmmrzdEx[,10]> PPBP <gg>
## 5 CD8+_transitional_effector_GZMK_KLRB1… <SmmrzdEx[,10]> TYROBP <gg>
## 6 HSC_lymphoid_myeloid_like <SmmrzdEx[,10]> GABPB1-A… <gg>
## 7 MAIT <SmmrzdEx[,10]> MT-CO1 <gg>
## 8 NK_cells <SmmrzdEx[,10]> MT-CO1 <gg>
## 9 NK_cycling <SmmrzdEx[,10]> CD3G <gg>
## 10 TCR_V_Delta_1 <SmmrzdEx[,10]> PRF1 <gg>
## 11 TCR_V_Delta_2 <SmmrzdEx[,9]> MT-CO1 <gg>
## 12 T_cell:CD8+_other <SmmrzdEx[,10]> CTSW <gg>
## 13 limphoid_myeloid_like_HLA_CD74 <SmmrzdEx[,4]> GNLY <gg>
## # ℹ abbreviated name: ¹grouped_summarized_experiment
pseudo_bulk_nested |> pull(plot) ## [[1]]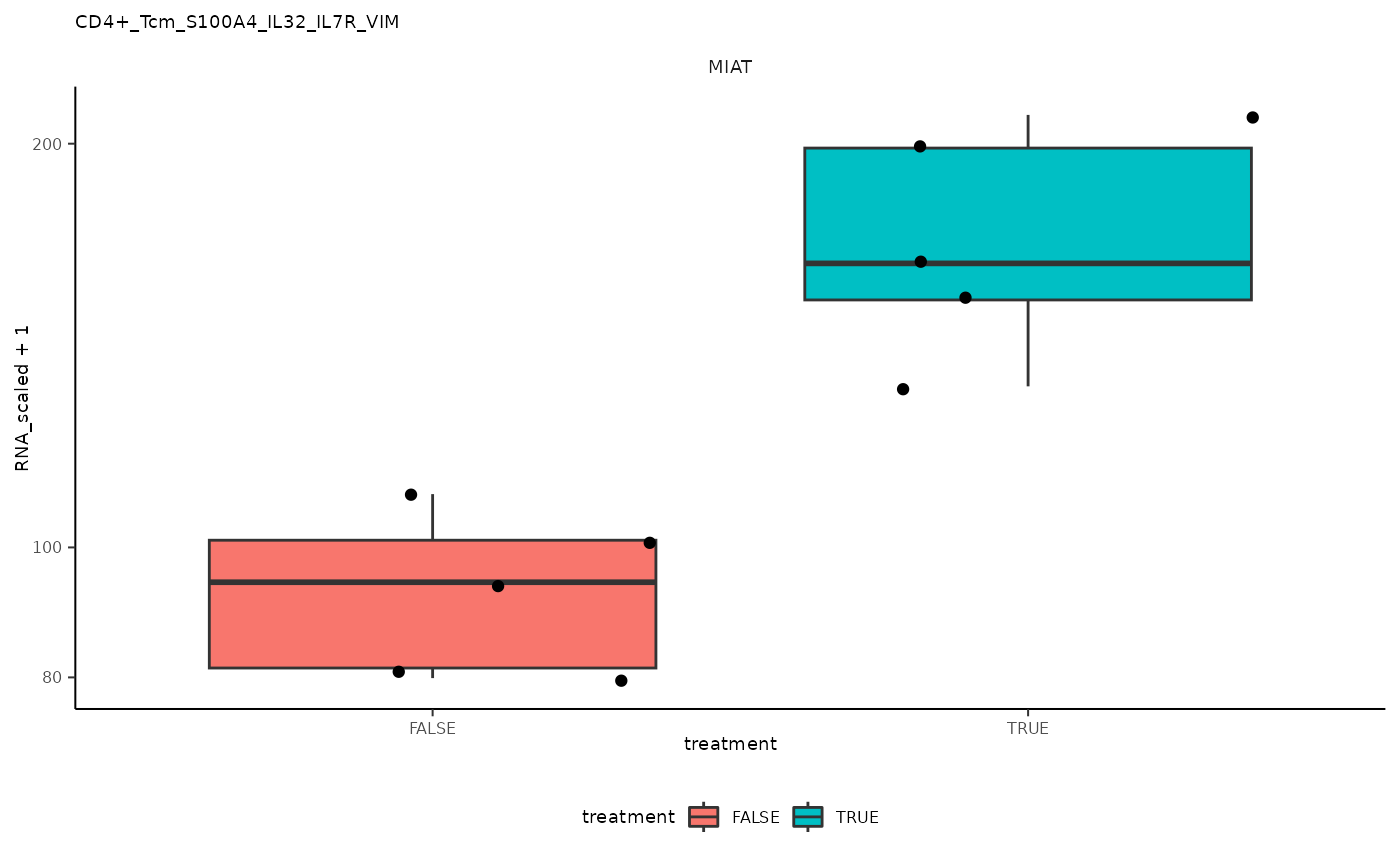
##
## [[2]]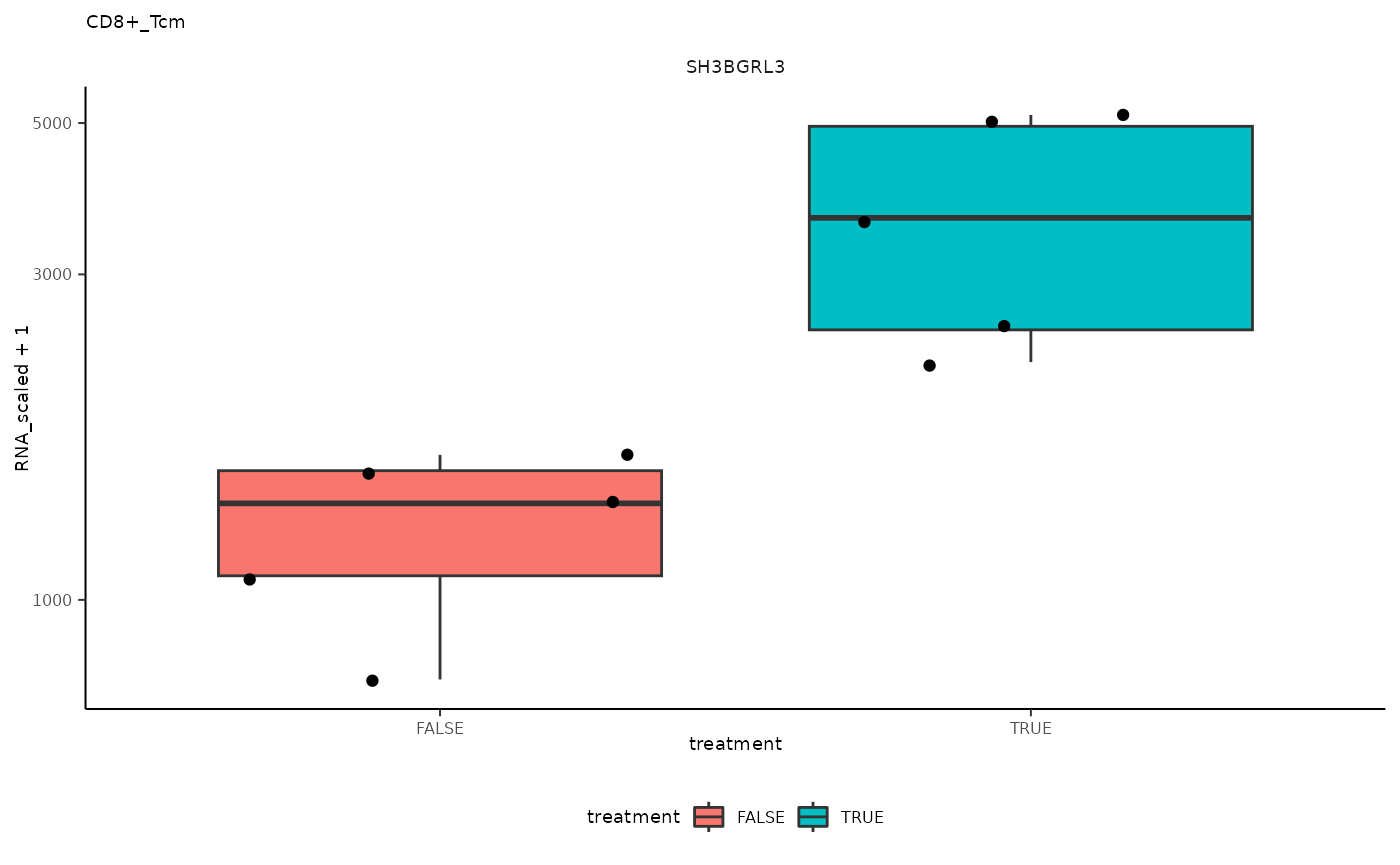
##
## [[3]]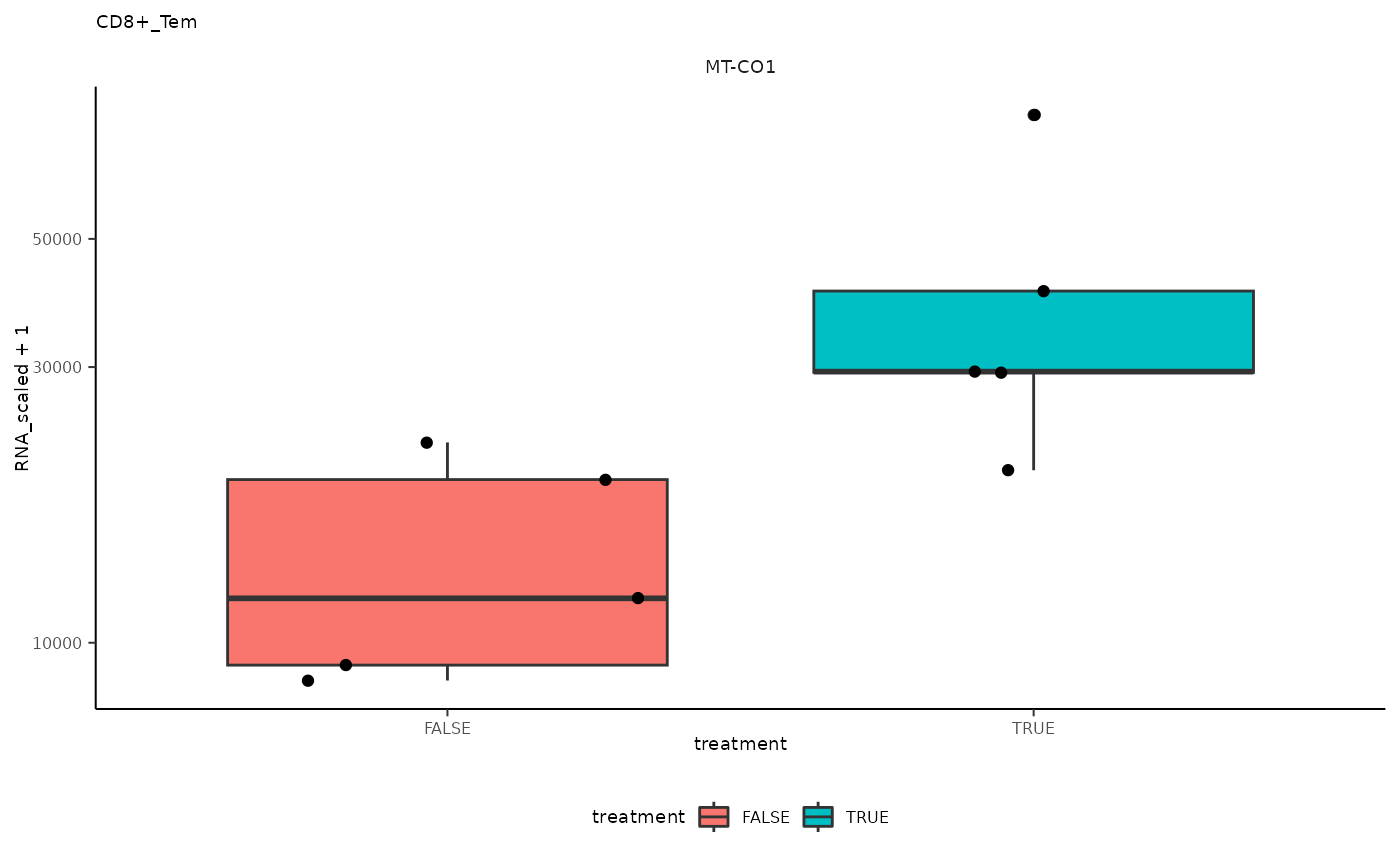
##
## [[4]]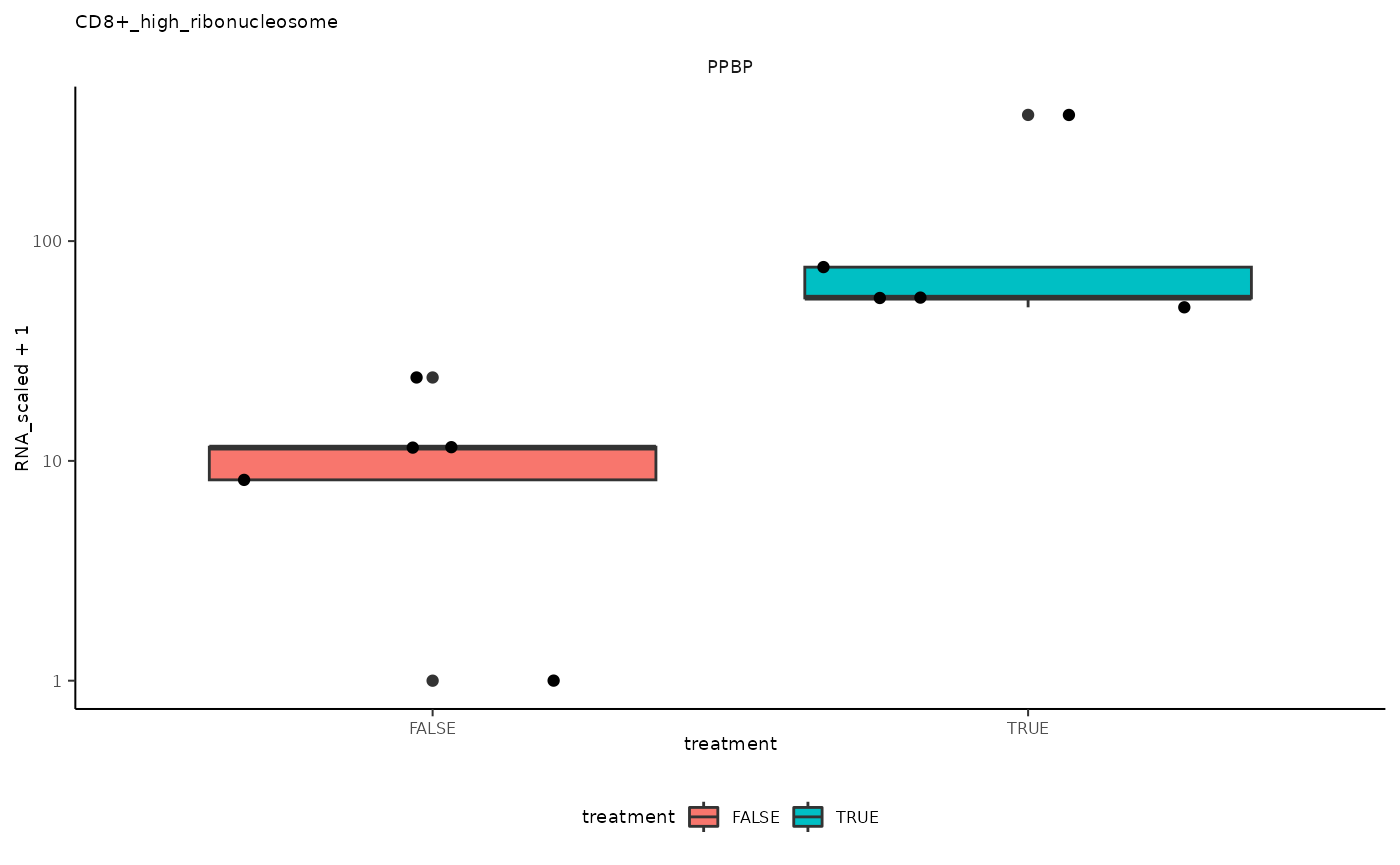
##
## [[5]]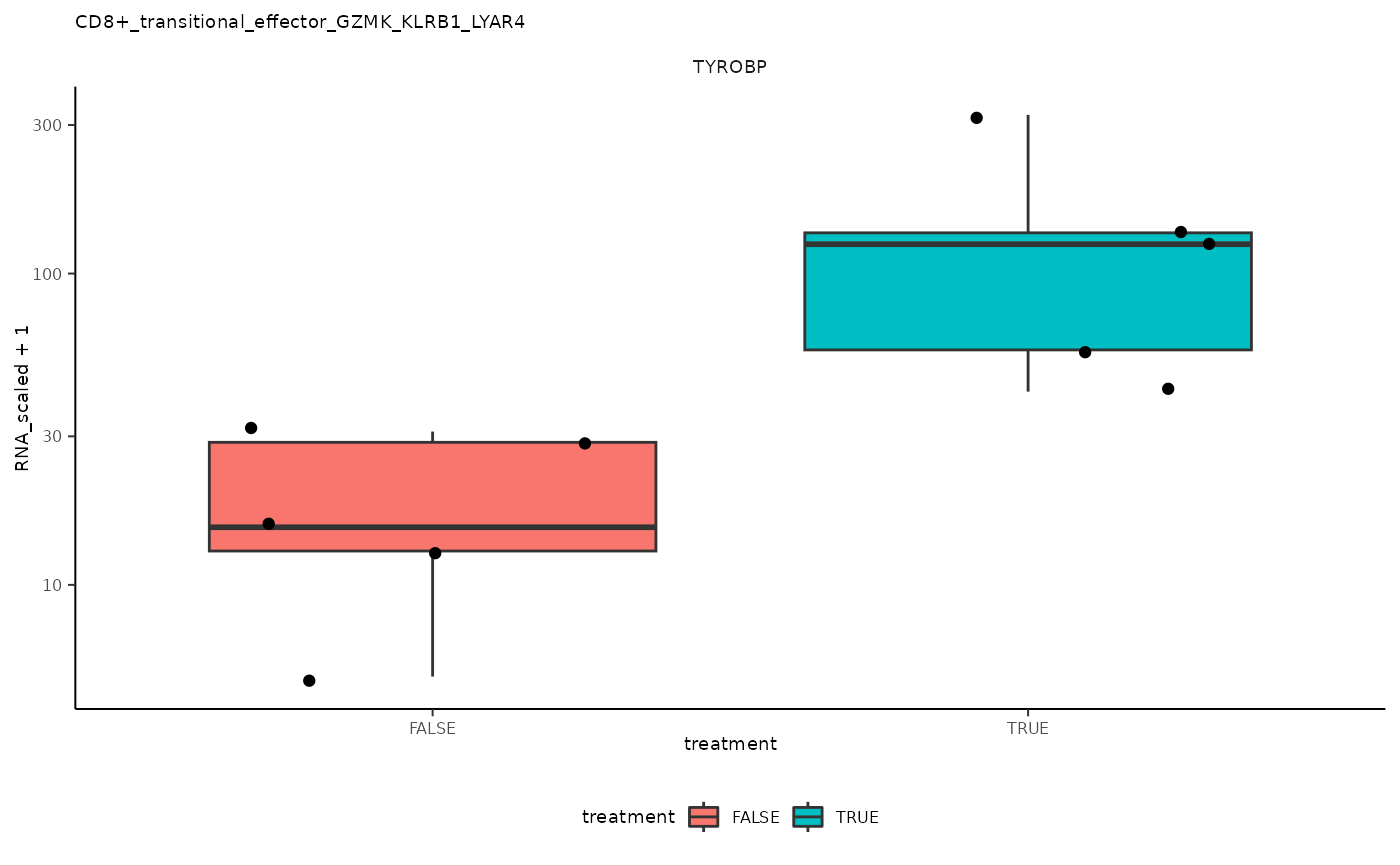
##
## [[6]]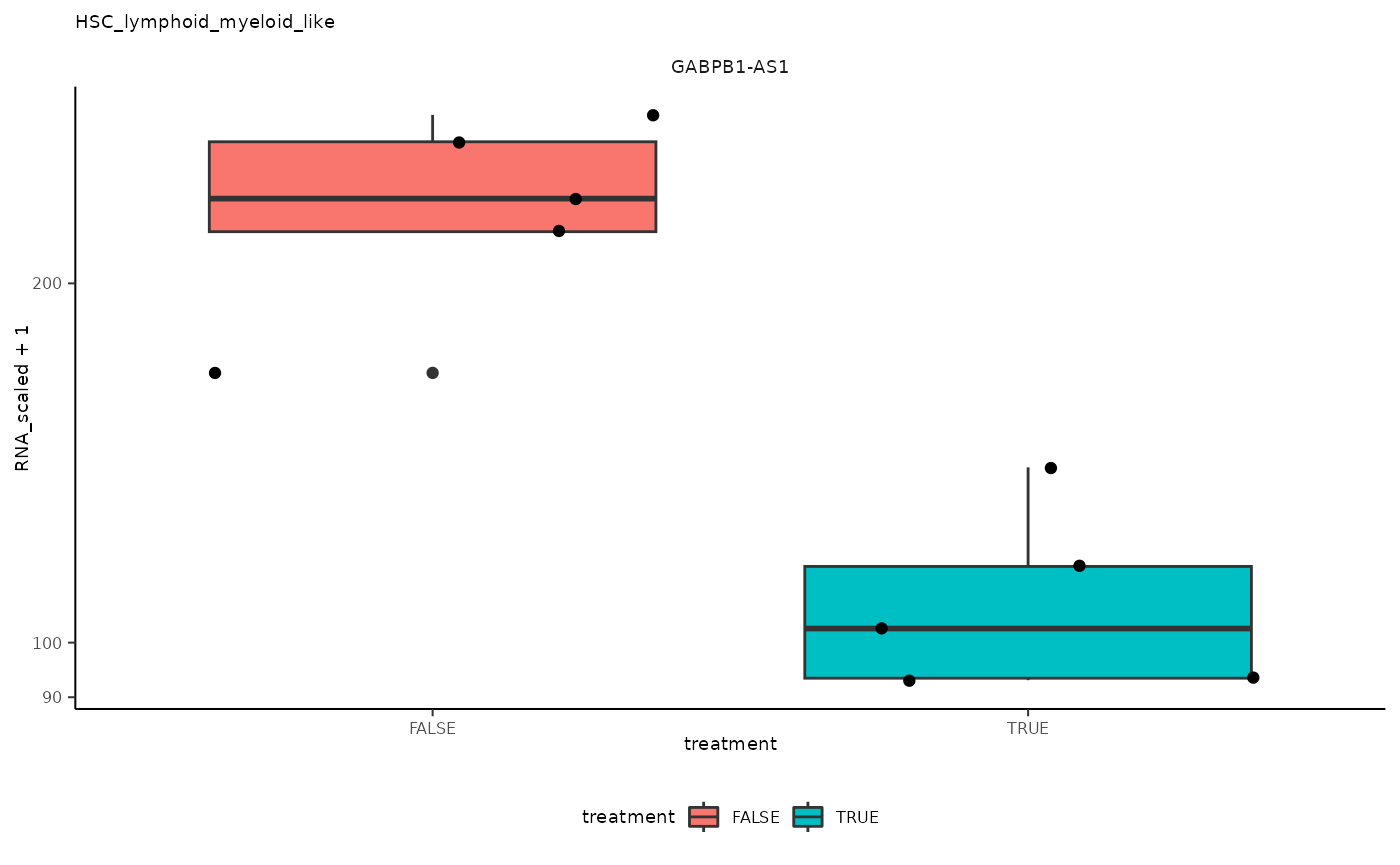
##
## [[7]]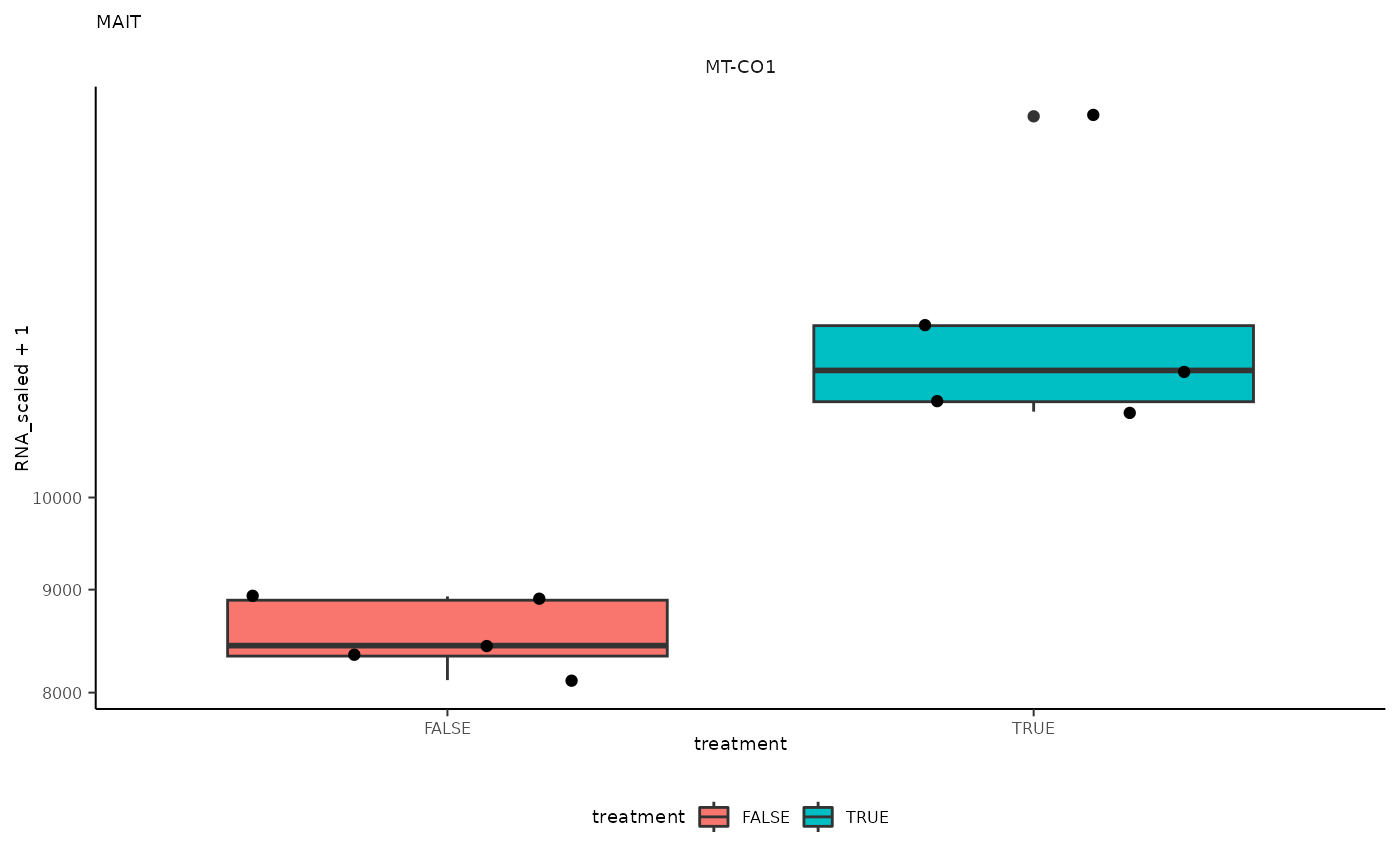
##
## [[8]]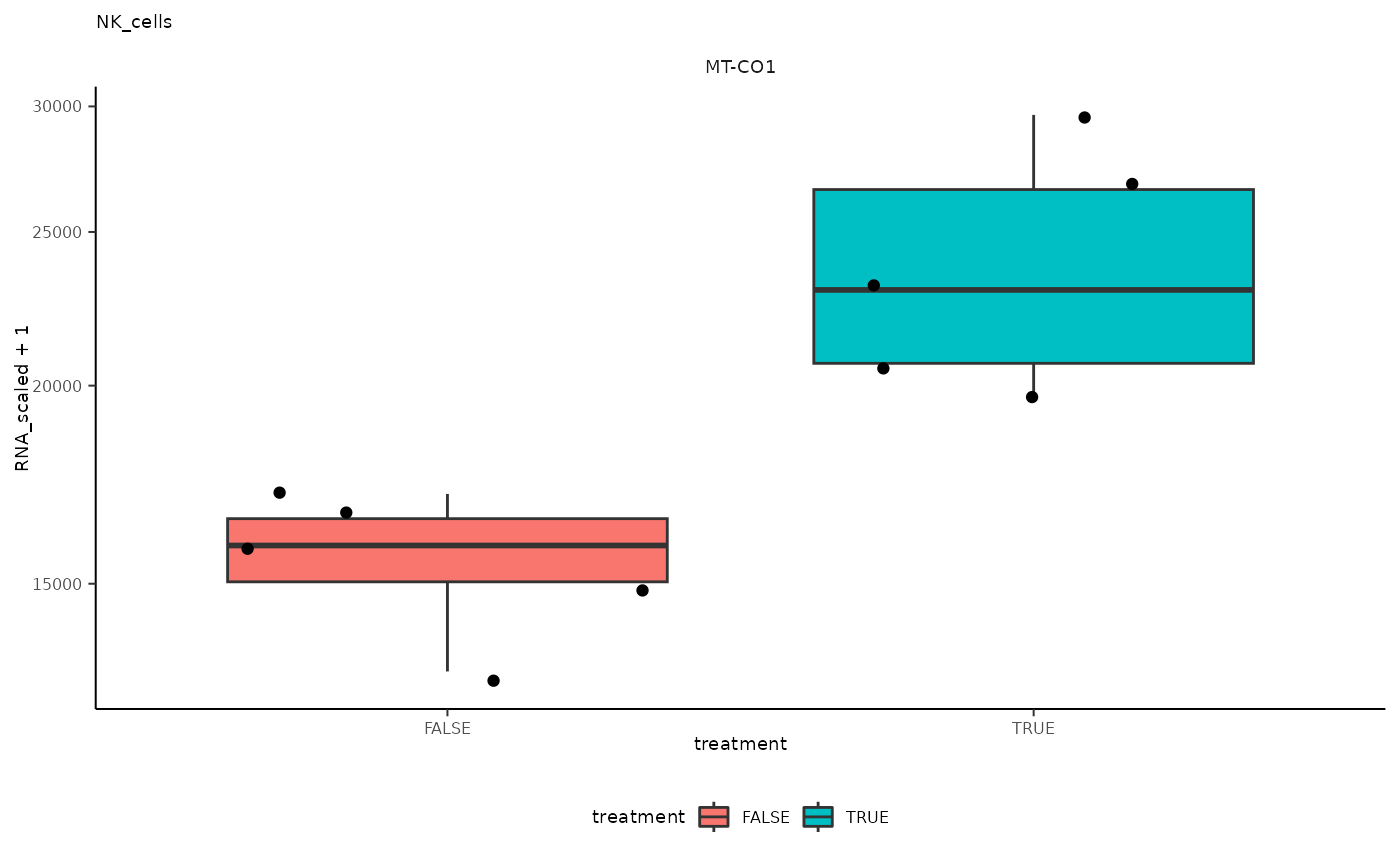
##
## [[9]]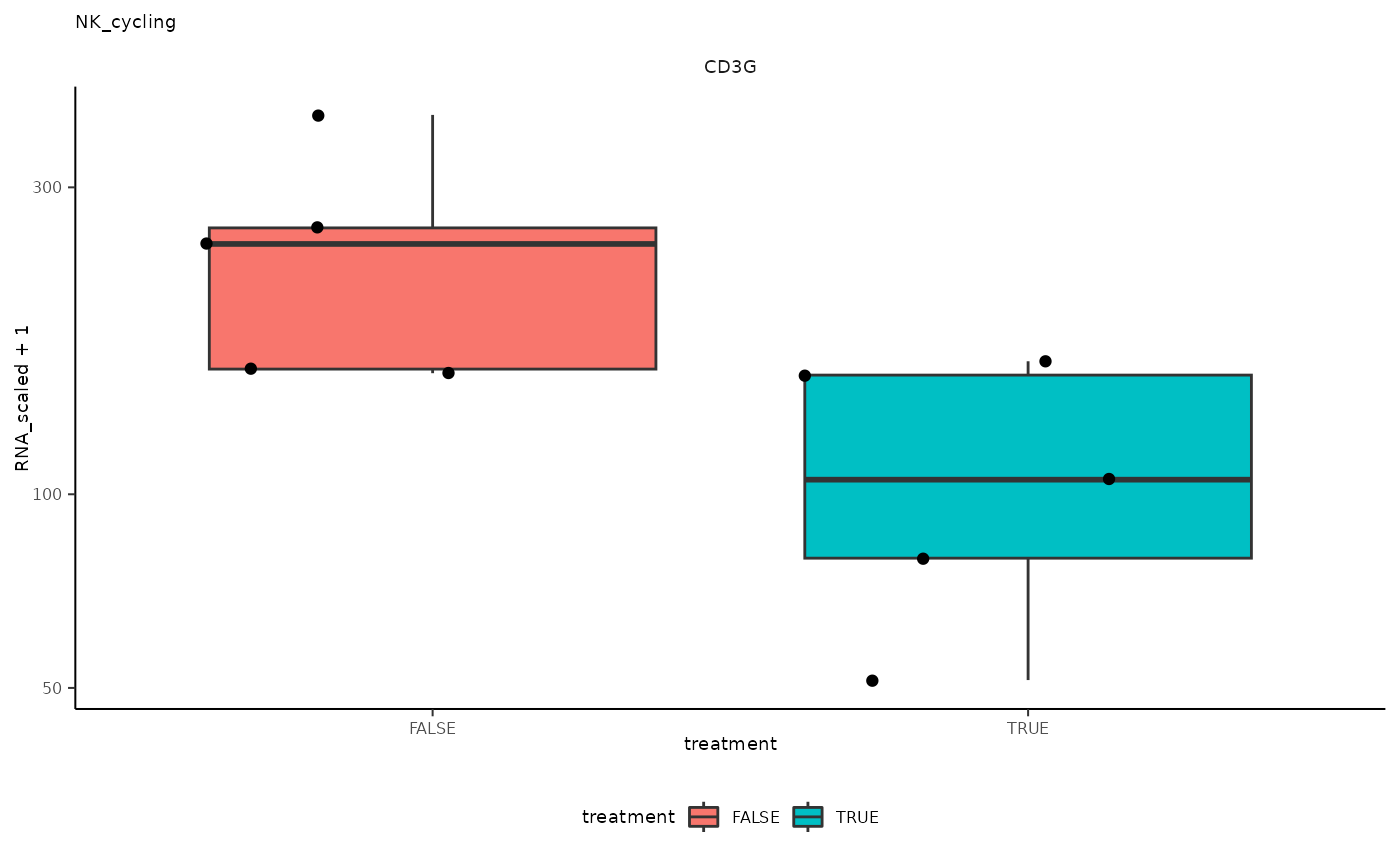
##
## [[10]]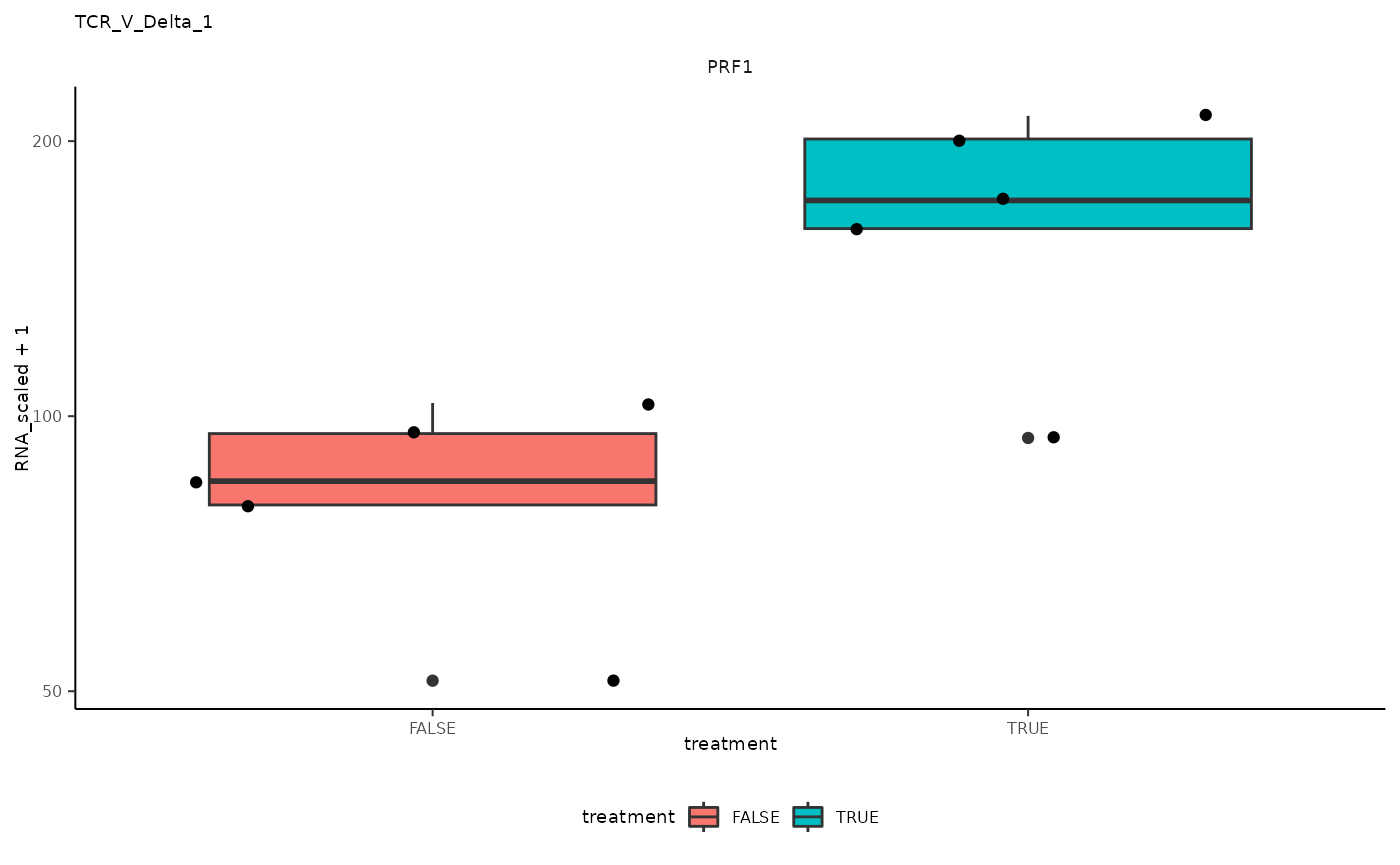
##
## [[11]]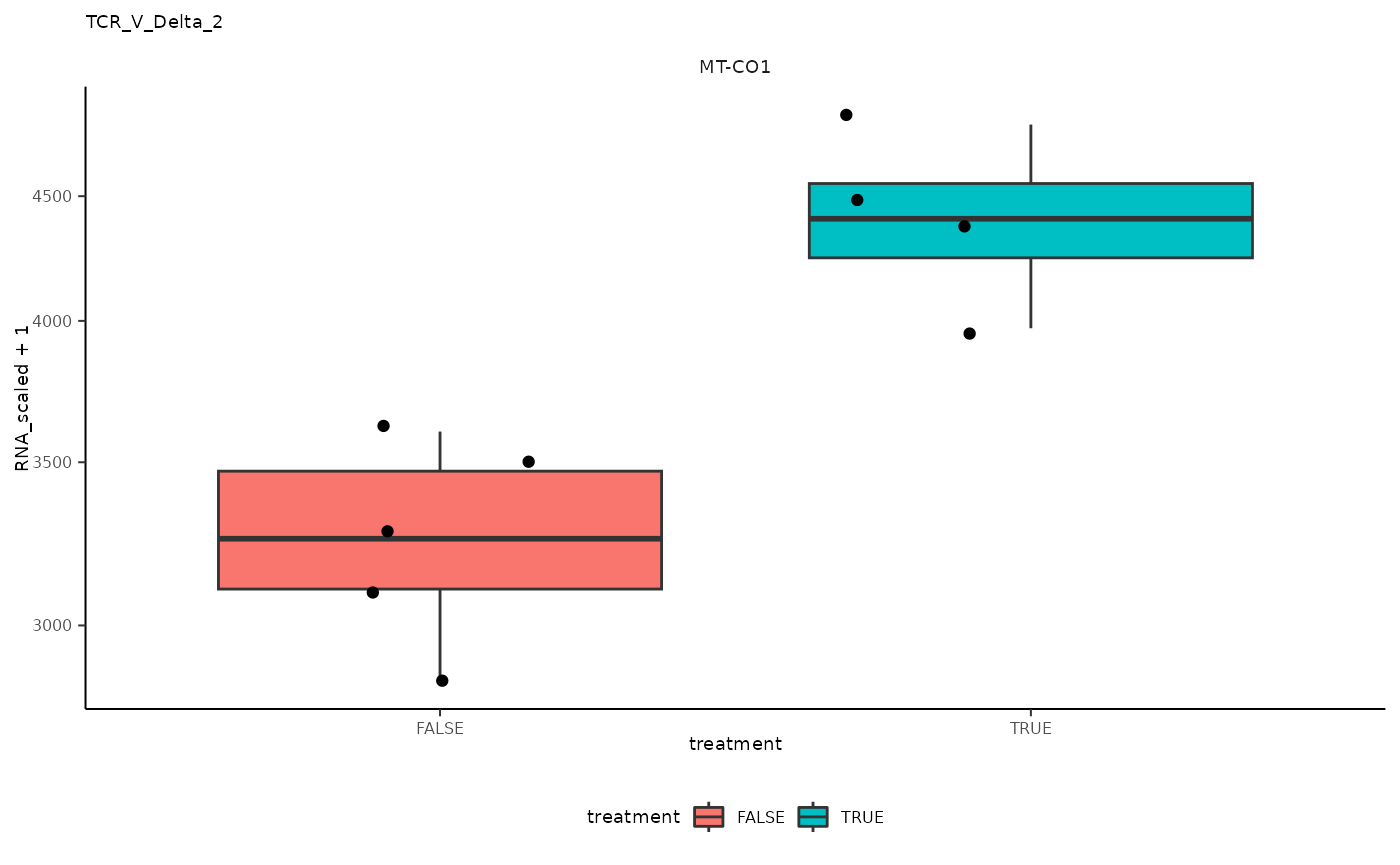
##
## [[12]]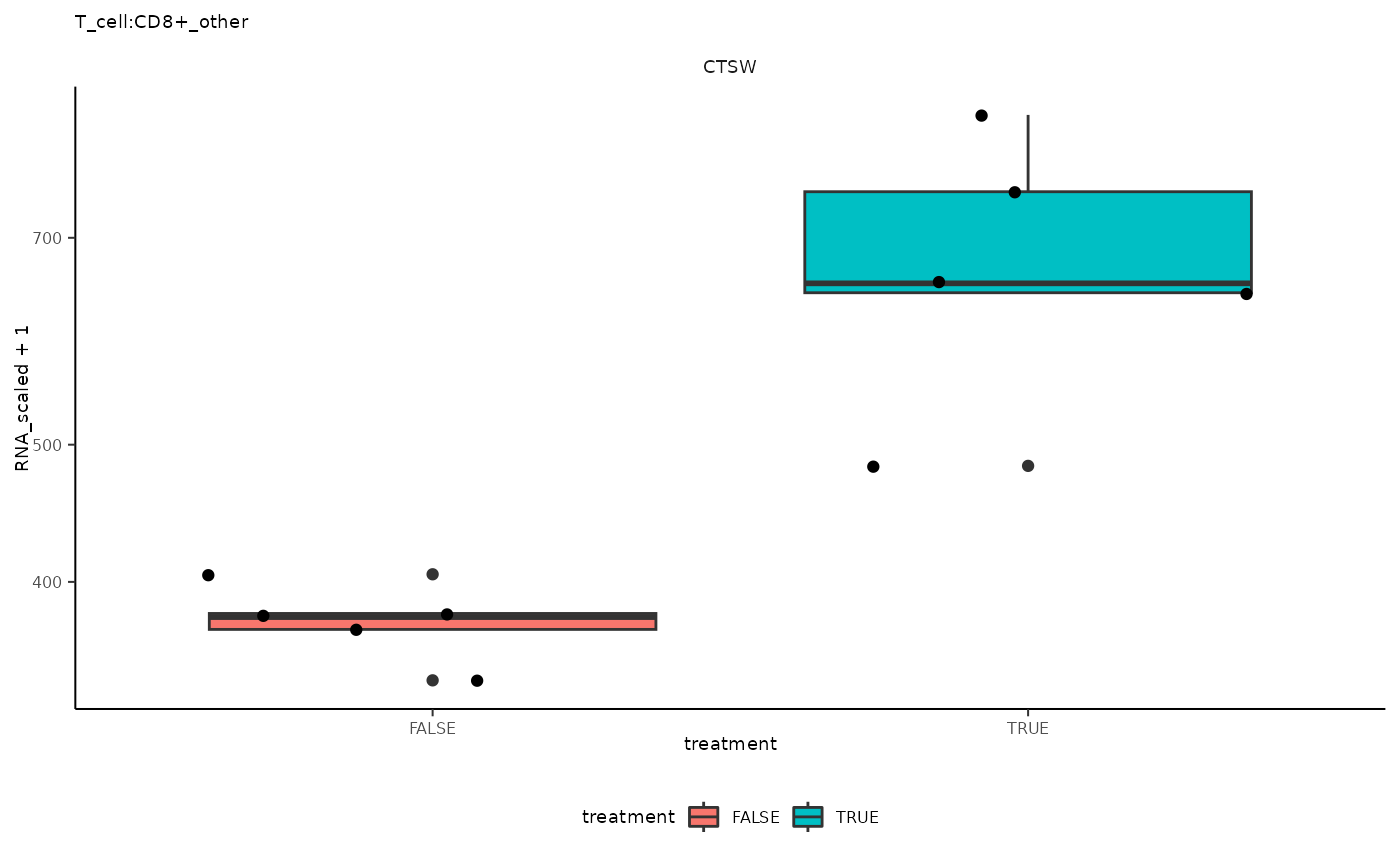
##
## [[13]]
Session Information
## R version 4.2.1 (2022-06-23)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] tidySummarizedExperiment_1.9.1 SummarizedExperiment_1.26.1
## [3] Biobase_2.56.0 GenomicRanges_1.48.0
## [5] GenomeInfoDb_1.32.4 IRanges_2.30.1
## [7] S4Vectors_0.34.0 BiocGenerics_0.42.0
## [9] MatrixGenerics_1.8.1 matrixStats_1.0.0
## [11] tidybulk_1.11.1 tidyr_1.3.0
## [13] glue_1.6.2 tidyseurat_0.5.9
## [15] ttservice_0.3.5 dittoSeq_1.8.1
## [17] colorspace_2.1-0 dplyr_1.1.2
## [19] ggplot2_3.4.2 SeuratObject_4.1.3
## [21] Seurat_4.3.0 purrr_1.0.1
##
## loaded via a namespace (and not attached):
## [1] utf8_1.2.3
## [2] spatstat.explore_3.2-1
## [3] reticulate_1.29
## [4] tidyselect_1.2.0
## [5] RSQLite_2.3.1
## [6] AnnotationDbi_1.58.0
## [7] htmlwidgets_1.6.2
## [8] grid_4.2.1
## [9] BiocParallel_1.30.4
## [10] Rtsne_0.16
## [11] munsell_0.5.0
## [12] codetools_0.2-19
## [13] ragg_1.2.5
## [14] ica_1.0-3
## [15] preprocessCore_1.58.0
## [16] future_1.32.0
## [17] miniUI_0.1.1.1
## [18] withr_2.5.0
## [19] spatstat.random_3.1-5
## [20] progressr_0.13.0
## [21] highr_0.10
## [22] knitr_1.43
## [23] SingleCellExperiment_1.18.1
## [24] ROCR_1.0-11
## [25] tensor_1.5
## [26] listenv_0.9.0
## [27] labeling_0.4.2
## [28] GenomeInfoDbData_1.2.8
## [29] polyclip_1.10-4
## [30] bit64_4.0.5
## [31] farver_2.1.1
## [32] pheatmap_1.0.12
## [33] rprojroot_2.0.3
## [34] parallelly_1.36.0
## [35] vctrs_0.6.2
## [36] generics_0.1.3
## [37] xfun_0.39
## [38] R6_2.5.1
## [39] locfit_1.5-9.7
## [40] bitops_1.0-7
## [41] spatstat.utils_3.0-3
## [42] cachem_1.0.8
## [43] DelayedArray_0.22.0
## [44] promises_1.2.0.1
## [45] scales_1.2.1
## [46] gtable_0.3.3
## [47] RMedicine2023tidytranscriptomics_0.15.0
## [48] globals_0.16.2
## [49] goftest_1.2-3
## [50] rlang_1.1.1
## [51] genefilter_1.78.0
## [52] systemfonts_1.0.4
## [53] splines_4.2.1
## [54] lazyeval_0.2.2
## [55] spatstat.geom_3.2-1
## [56] BiocManager_1.30.20
## [57] yaml_2.3.7
## [58] reshape2_1.4.4
## [59] abind_1.4-5
## [60] crosstalk_1.2.0
## [61] httpuv_1.6.11
## [62] tools_4.2.1
## [63] ellipsis_0.3.2
## [64] jquerylib_0.1.4
## [65] RColorBrewer_1.1-3
## [66] ggridges_0.5.4
## [67] Rcpp_1.0.10
## [68] plyr_1.8.8
## [69] zlibbioc_1.42.0
## [70] RCurl_1.98-1.12
## [71] deldir_1.0-9
## [72] pbapply_1.7-0
## [73] cowplot_1.1.1
## [74] zoo_1.8-12
## [75] ggrepel_0.9.3
## [76] cluster_2.1.4
## [77] fs_1.6.2
## [78] magrittr_2.0.3
## [79] data.table_1.14.8
## [80] scattermore_1.1
## [81] lmtest_0.9-40
## [82] RANN_2.6.1
## [83] fitdistrplus_1.1-11
## [84] hms_1.1.3
## [85] patchwork_1.1.2
## [86] mime_0.12
## [87] evaluate_0.21
## [88] xtable_1.8-4
## [89] XML_3.99-0.14
## [90] gridExtra_2.3
## [91] compiler_4.2.1
## [92] tibble_3.2.1
## [93] crayon_1.5.2
## [94] KernSmooth_2.23-21
## [95] htmltools_0.5.5
## [96] later_1.3.1
## [97] tzdb_0.4.0
## [98] geneplotter_1.74.0
## [99] DBI_1.1.3
## [100] MASS_7.3-60
## [101] Matrix_1.5-4.1
## [102] readr_2.1.4
## [103] cli_3.6.1
## [104] parallel_4.2.1
## [105] igraph_1.4.3
## [106] pkgconfig_2.0.3
## [107] pkgdown_2.0.7
## [108] sp_1.6-1
## [109] plotly_4.10.2
## [110] spatstat.sparse_3.0-1
## [111] annotate_1.74.0
## [112] bslib_0.4.2
## [113] XVector_0.36.0
## [114] stringr_1.5.0
## [115] digest_0.6.31
## [116] sctransform_0.3.5
## [117] RcppAnnoy_0.0.20
## [118] Biostrings_2.64.1
## [119] spatstat.data_3.0-1
## [120] rmarkdown_2.22
## [121] leiden_0.4.3
## [122] uwot_0.1.14
## [123] edgeR_3.38.4
## [124] shiny_1.7.4
## [125] lifecycle_1.0.3
## [126] nlme_3.1-162
## [127] jsonlite_1.8.5
## [128] desc_1.4.2
## [129] viridisLite_0.4.2
## [130] limma_3.52.4
## [131] fansi_1.0.4
## [132] pillar_1.9.0
## [133] lattice_0.21-8
## [134] KEGGREST_1.36.3
## [135] fastmap_1.1.1
## [136] httr_1.4.6
## [137] survival_3.5-5
## [138] png_0.1-8
## [139] bit_4.0.5
## [140] stringi_1.7.12
## [141] sass_0.4.6
## [142] blob_1.2.4
## [143] textshaping_0.3.6
## [144] DESeq2_1.36.0
## [145] memoise_2.0.1
## [146] irlba_2.3.5.1
## [147] future.apply_1.11.0References